

Hypomorphic Mutations in TONSL Cause SPONASTRIME Dysplasia
- PMID: 30773278
- PMCID: PMC6407524
- DOI: 10.1016/j.ajhg.2019.01.009
Hypomorphic Mutations in TONSL Cause SPONASTRIME Dysplasia
Abstract
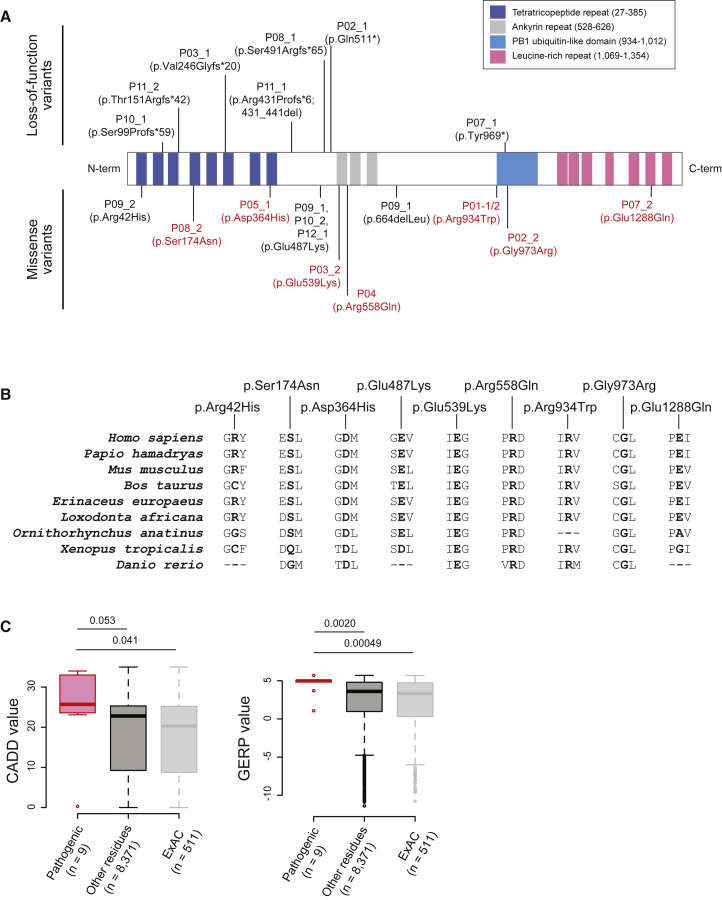
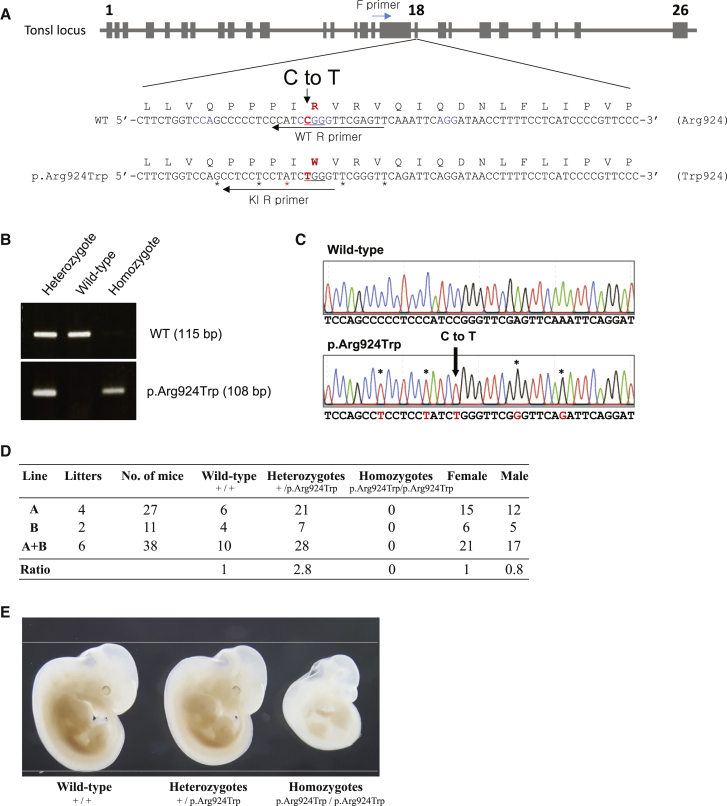
SPONASTRIME dysplasia is a rare, recessive skeletal dysplasia characterized by short stature, facial dysmorphism, and aberrant radiographic findings of the spine and long bone metaphysis. No causative genetic alterations for SPONASTRIME dysplasia have yet been determined. Using whole-exome sequencing (WES), we identified bi-allelic TONSL mutations in 10 of 13 individuals with SPONASTRIME dysplasia. TONSL is a multi-domain scaffold protein that interacts with DNA replication and repair factors and which plays critical roles in resistance to replication stress and the maintenance of genome integrity. We show here that cellular defects in dermal fibroblasts from affected individuals are complemented by the expression of wild-type TONSL. In addition, in vitro cell-based assays and in silico analyses of TONSL structure support the pathogenicity of those TONSL variants. Intriguingly, a knock-in (KI) Tonsl mouse model leads to embryonic lethality, implying the physiological importance of TONSL. Overall, these findings indicate that genetic variants resulting in reduced function of TONSL cause SPONASTRIME dysplasia and highlight the importance of TONSL in embryonic development and postnatal growth.
Keywords: DNA repair; DNA replication; SPONASTRIME dysplasia; TONSL; rare genetic diseases; short stature; skeletal dysplasia; whole-exome sequencing.
Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures



References
-
- Fanconi S., Issler C., Giedion A., Prader A. The SPONASTRIME dysplasia: Familial short-limb dwarfism with saddle nose, spinal alterations and metaphyseal striation. Report of 4 siblings. Helv. Paediatr. Acta. 1983;38:267–280. - PubMed
-
- Lachman R.S., Stoss H., Spranger J. Sponastrime dysplasia. A radiologic-pathologic correlation. Pediatr. Radiol. 1989;19:417–424. - PubMed
-
- Langer L.O., Jr., Beals R.K., LaFranchi S., Scott C.I., Jr., Sockalosky J.J. Sponastrime dysplasia: Five new cases and review of nine previously published cases. Am. J. Med. Genet. 1996;63:20–27. - PubMed
-
- Masuno M., Nishimura G., Adachi M., Hotsubo T., Tachibana K., Makita Y., Imaizumi K., Kuroki Y. SPONASTRIME dysplasia: Report on a female patient with severe skeletal changes. Am. J. Med. Genet. 1996;66:429–432. - PubMed
-
- Langer L.O., Jr., Beals R.K., Scott C.I., Jr. Sponastrime dysplasia: Diagnostic criteria based on five new and six previously published cases. Pediatr. Radiol. 1997;27:409–414. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
