

DriverML: a machine learning algorithm for identifying driver genes in cancer sequencing studies
- PMID: 30773592
- PMCID: PMC6486576
- DOI: 10.1093/nar/gkz096
DriverML: a machine learning algorithm for identifying driver genes in cancer sequencing studies
Erratum in
-
Corrigendum to article "DriverML: a machine learning algorithm for identifying driver genes in cancer sequencing studies''.Nucleic Acids Res. 2021 Apr 19;49(7):4196. doi: 10.1093/nar/gkab193. Nucleic Acids Res. 2021. PMID: 33744935 Free PMC article. No abstract available.
Abstract
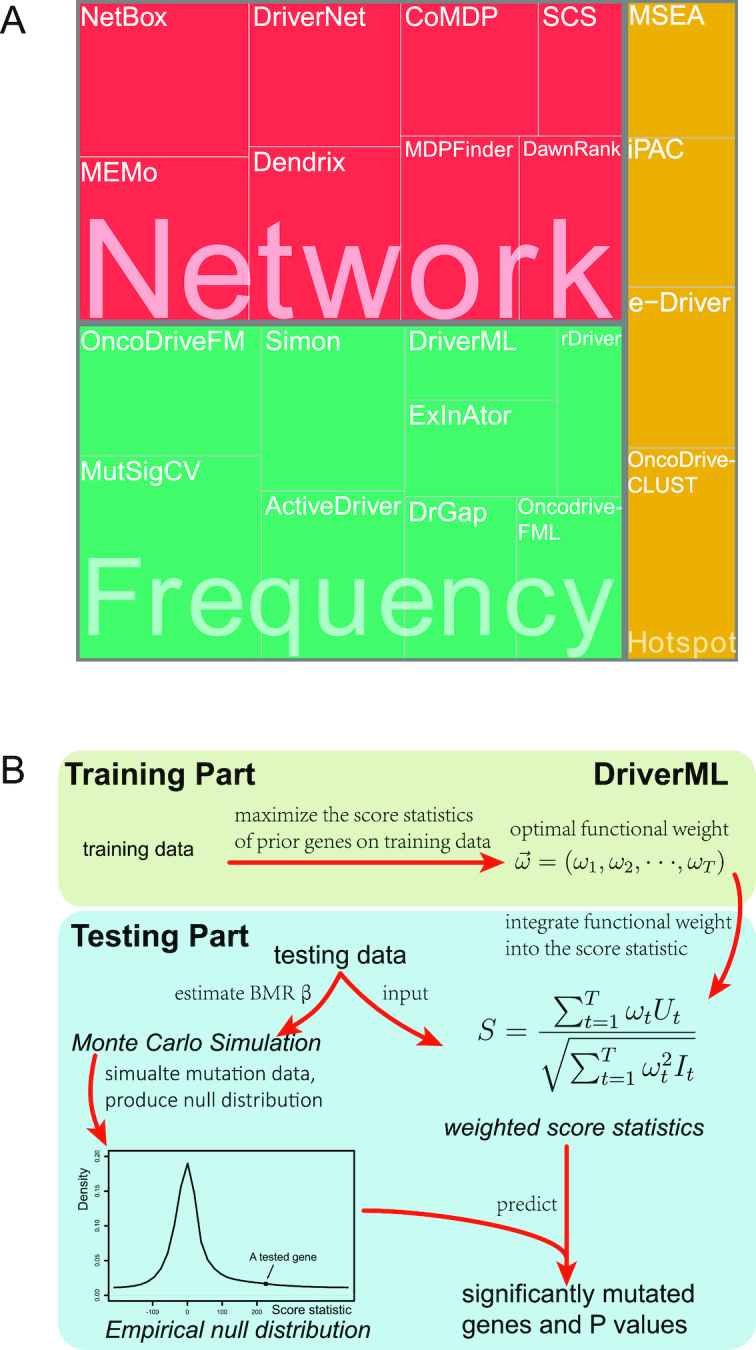
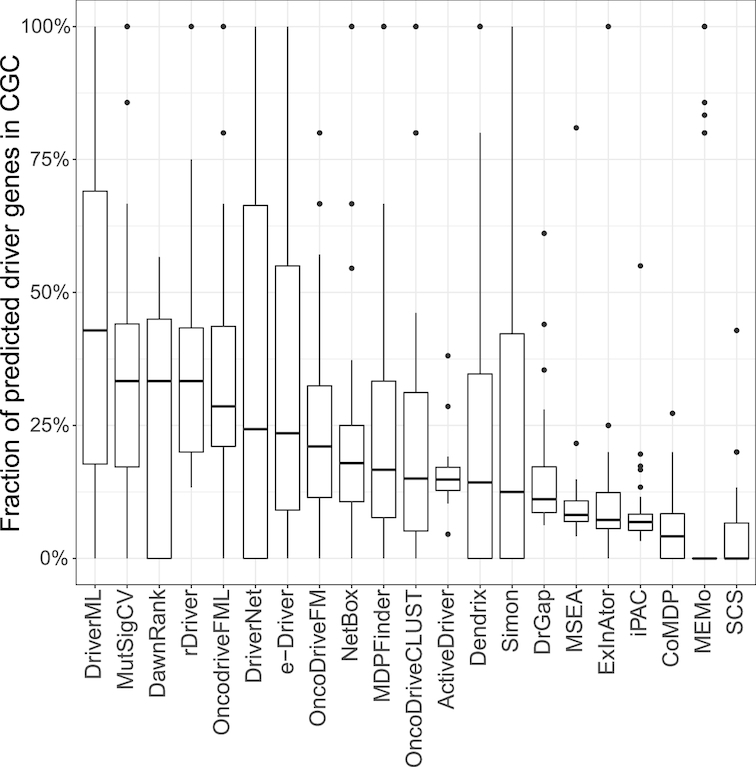
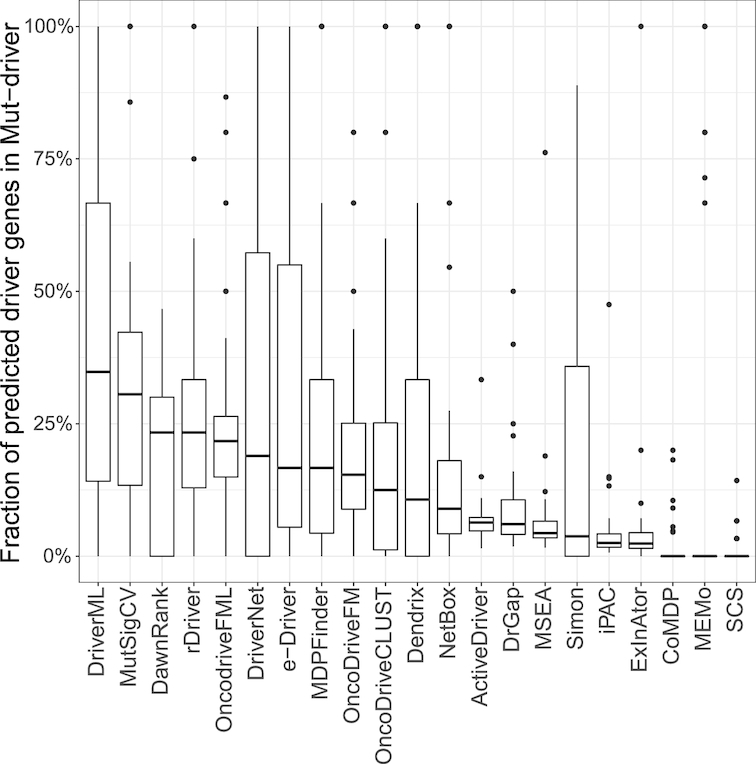
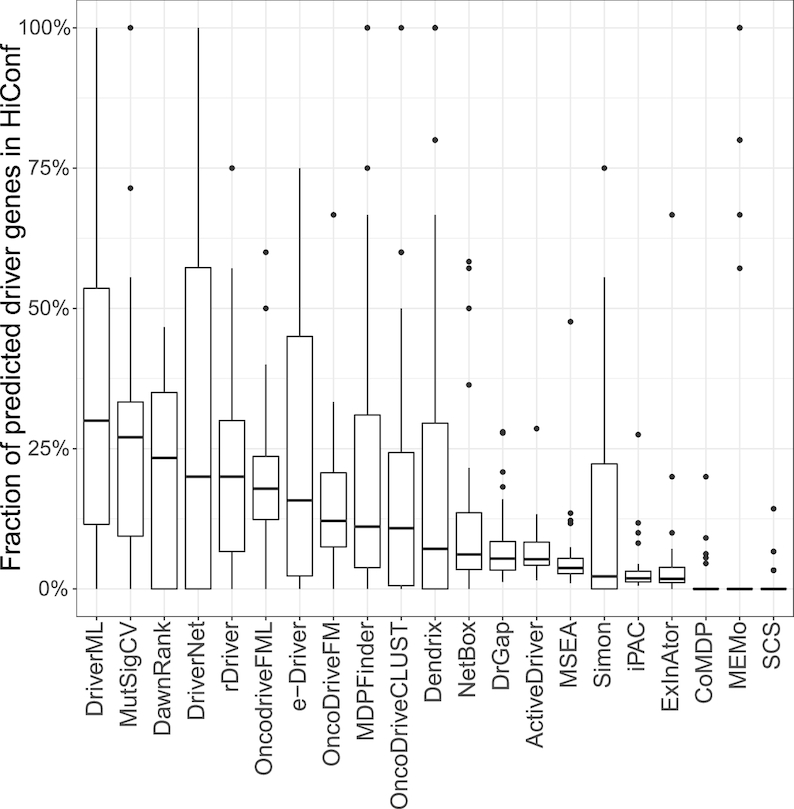
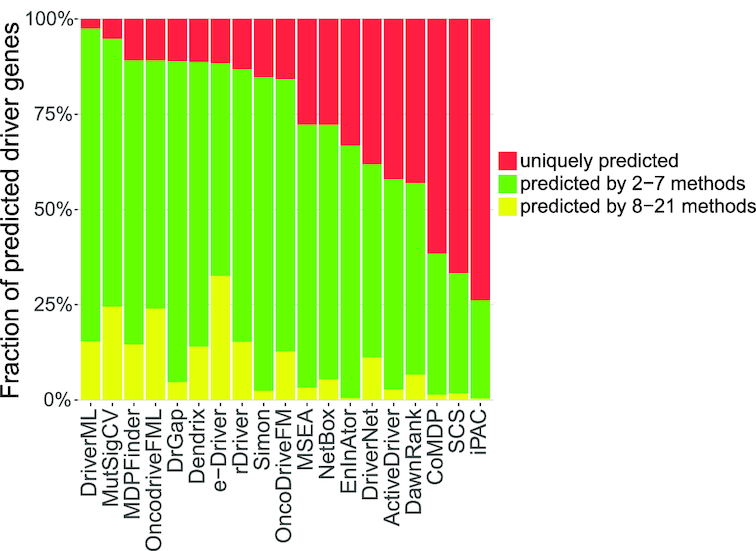
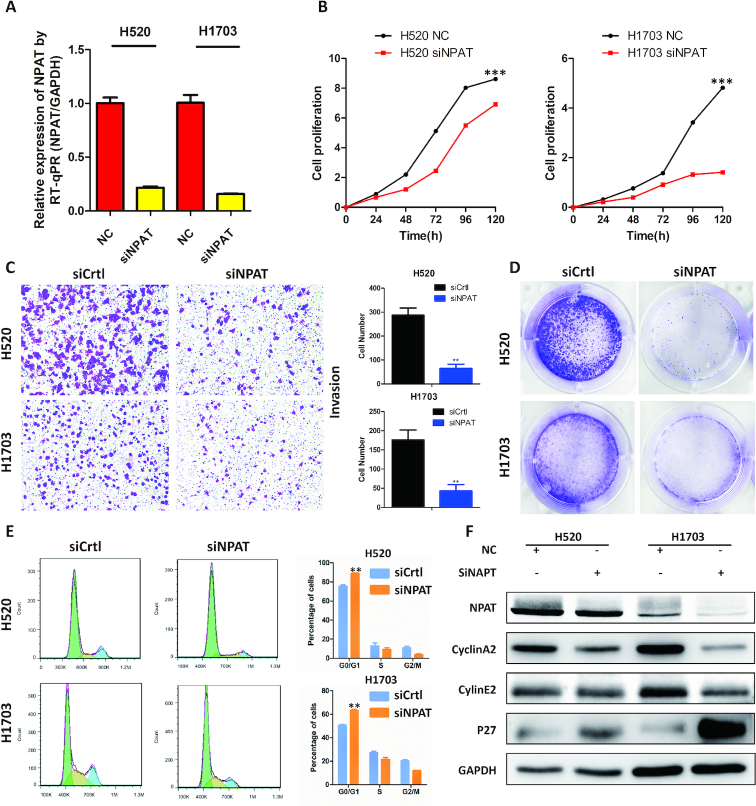
Although rapid progress has been made in computational approaches for prioritizing cancer driver genes, research is far from achieving the ultimate goal of discovering a complete catalog of genes truly associated with cancer. Driver gene lists predicted from these computational tools lack consistency and are prone to false positives. Here, we developed an approach (DriverML) integrating Rao's score test and supervised machine learning to identify cancer driver genes. The weight parameters in the score statistics quantified the functional impacts of mutations on the protein. To obtain optimized weight parameters, the score statistics of prior driver genes were maximized on pan-cancer training data. We conducted rigorous and unbiased benchmark analysis and comparisons of DriverML with 20 other existing tools in 31 independent datasets from The Cancer Genome Atlas (TCGA). Our comprehensive evaluations demonstrated that DriverML was robust and powerful among various datasets and outperformed the other tools with a better balance of precision and sensitivity. In vitro cell-based assays further proved the validity of the DriverML prediction of novel driver genes. In summary, DriverML uses an innovative, machine learning-based approach to prioritize cancer driver genes and provides dramatic improvements over currently existing methods. Its source code is available at https://github.com/HelloYiHan/DriverML.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Meyerson M., Gabriel S., Getz G.. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010; 11:685–696. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
