

Cardiac-specific deletion of GCN5L1 restricts recovery from ischemia-reperfusion injury
- PMID: 30776374
- PMCID: PMC6486843
- DOI: 10.1016/j.yjmcc.2019.02.009
Cardiac-specific deletion of GCN5L1 restricts recovery from ischemia-reperfusion injury
Abstract
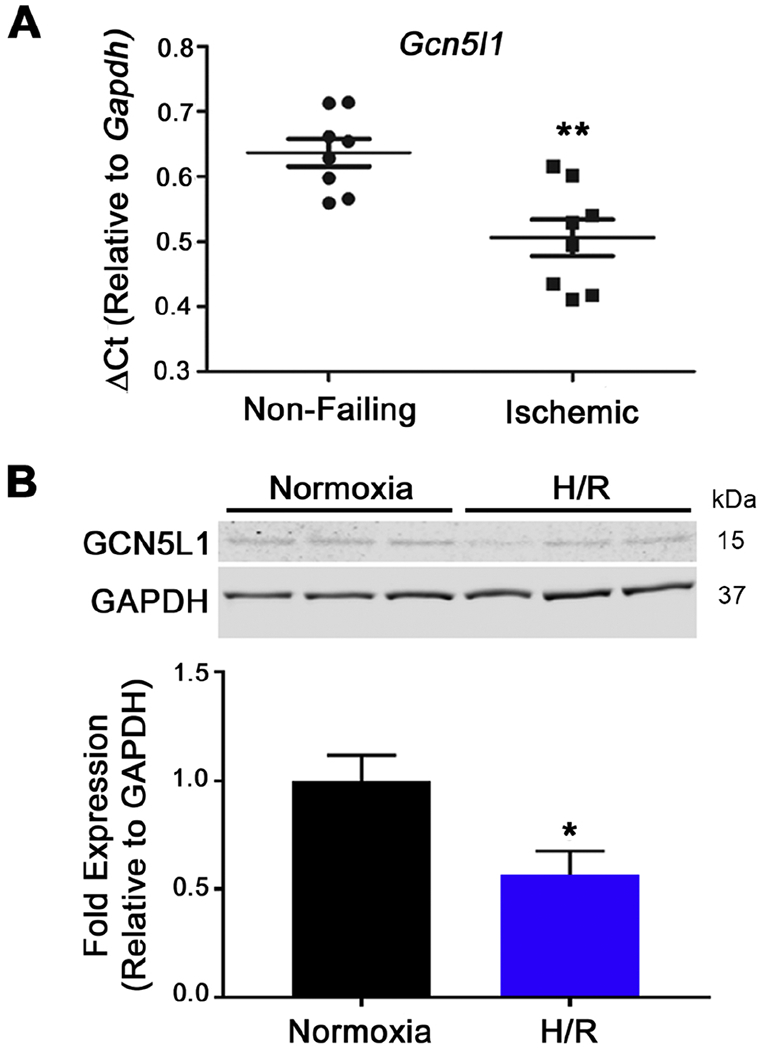
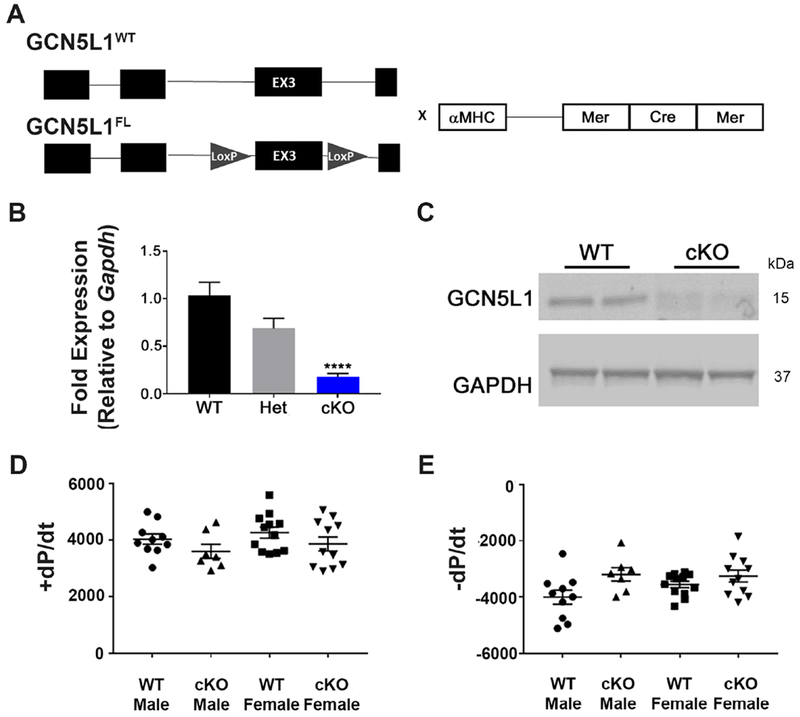
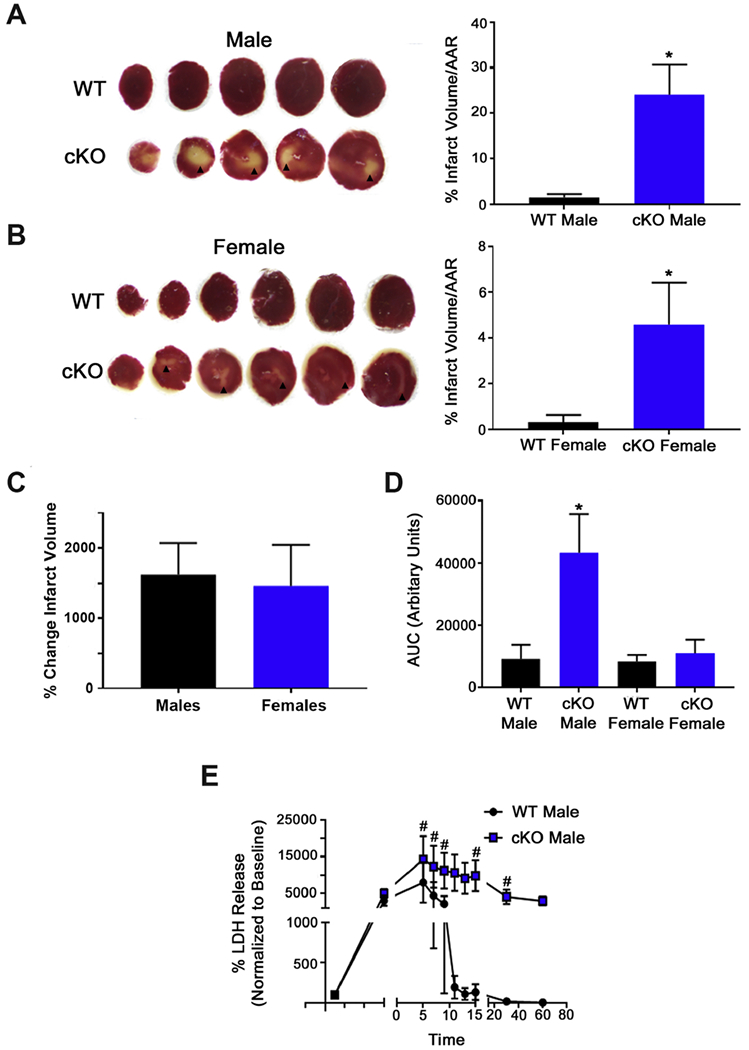
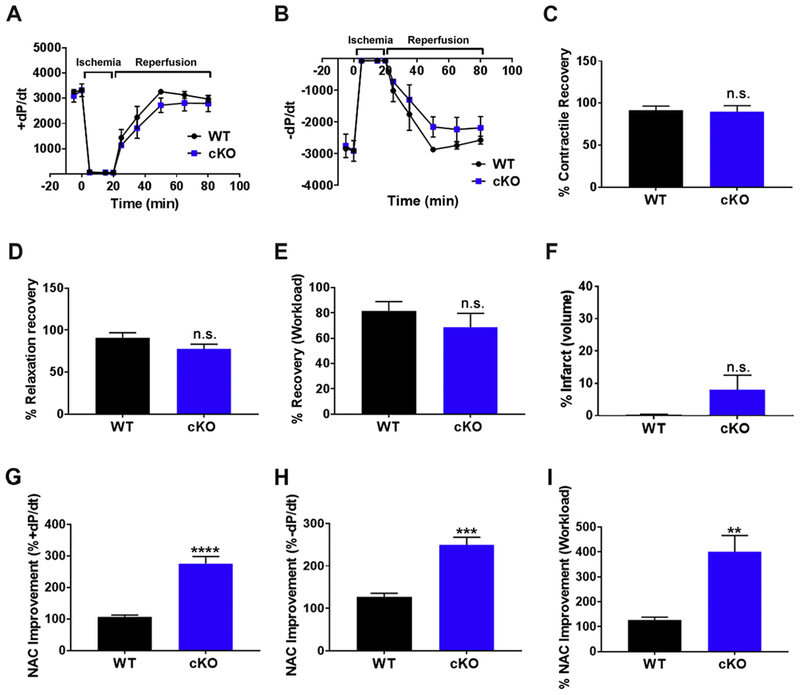
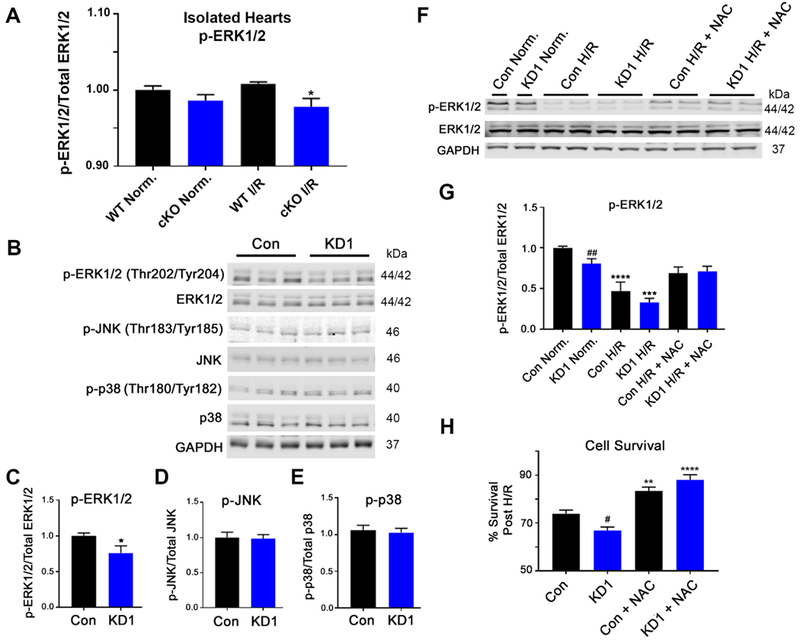
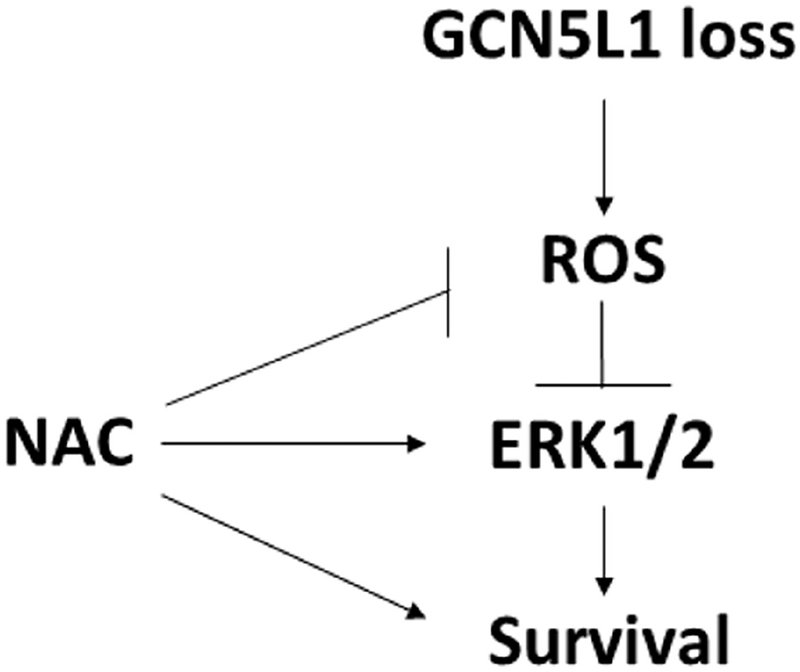
GCN5L1 regulates mitochondrial protein acetylation, cellular bioenergetics, reactive oxygen species (ROS) generation, and organelle positioning in a number of diverse cell types. However, the functional role of GCN5L1 in the heart is currently unknown. As many of the factors regulated by GCN5L1 play a major role in ischemia-reperfusion (I/R) injury, we sought to determine if GCN5L1 is an important nexus in the response to cardiac ischemic stress. Deletion of GCN5L1 in cardiomyocytes resulted in impaired myocardial post-ischemic function and increased infarct development in isolated work-performing hearts. GCN5L1 knockout hearts displayed hallmarks of ROS damage, and scavenging of ROS restored cardiac function and reduced infarct volume in vivo. GCN5L1 knockdown in cardiac-derived AC16 cells was associated with reduced activation of the pro-survival MAP kinase ERK1/2, which was also reversed by ROS scavenging, leading to restored cell viability. We therefore conclude that GCN5L1 activity provides an important protection against I/R induced, ROS-mediated damage in the ischemic heart.
Keywords: ERK1/2; Ex vivo working heart; GCN5L1; Ischemia reperfusion; Reactive oxygen species.
Copyright © 2019 Elsevier Ltd. All rights reserved.
Conflict of interest statement
CONFLICTS OF INTEREST
None to report.
Figures


References
-
- Brookes PS, Yoon Y, Robotham JL, Anders MW & Sheu S-S Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. - Cell Physiol 287, C817–C833 (2004). - PubMed
-
- Garcia-Rivas GJ & Torre-Amione G Abnormal mitochondrial function during ischemia reperfusion provides targets for pharmacological therapy. Methodist Debakey Cardiovasc. J 5, 2–7 (2009). - PubMed
-
- Shintani-Ishida K, Inui M & Yoshida K-I Ischemia-reperfusion induces myocardial infarction through mitochondrial Ca2+ overload. J. Mol. Cell. Cardiol 53, 233–239 (2012). - PubMed
-
- Alam MR, Baetz D & Ovize M Cyclophilin D and myocardial ischemia–reperfusion injury: A fresh perspective. J. Mol. Cell. Cardiol 78, 80–89 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
