

Long non-coding RNAs discriminate the stages and gene regulatory states of human humoral immune response
- PMID: 30778059
- PMCID: PMC6379396
- DOI: 10.1038/s41467-019-08679-z
Long non-coding RNAs discriminate the stages and gene regulatory states of human humoral immune response
Abstract
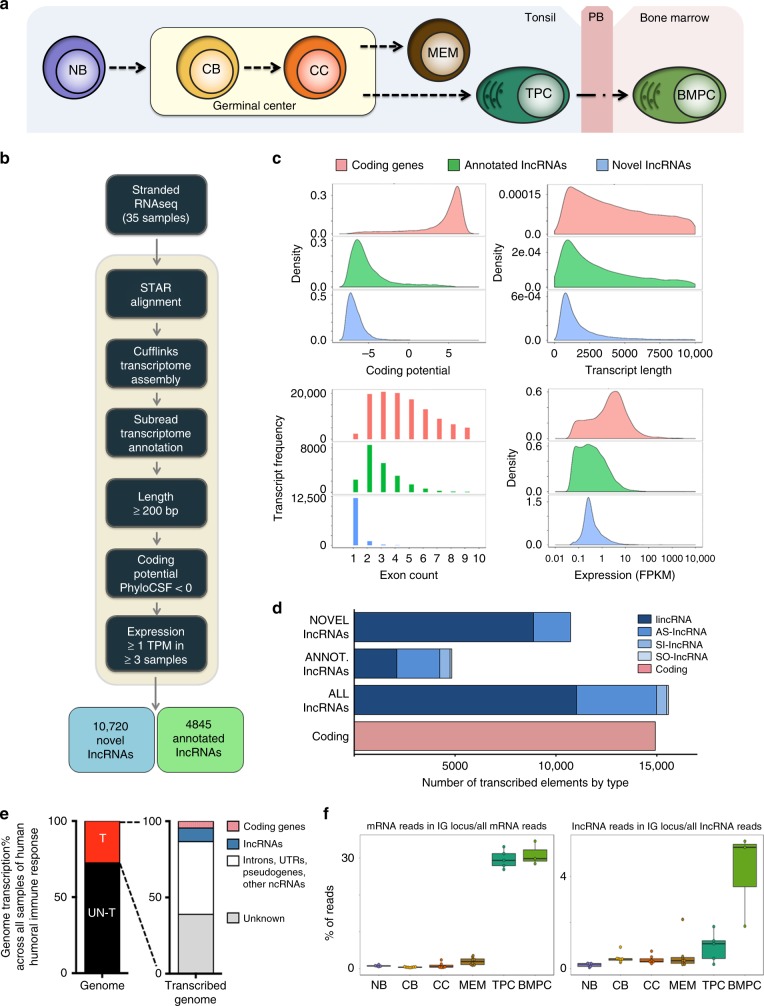
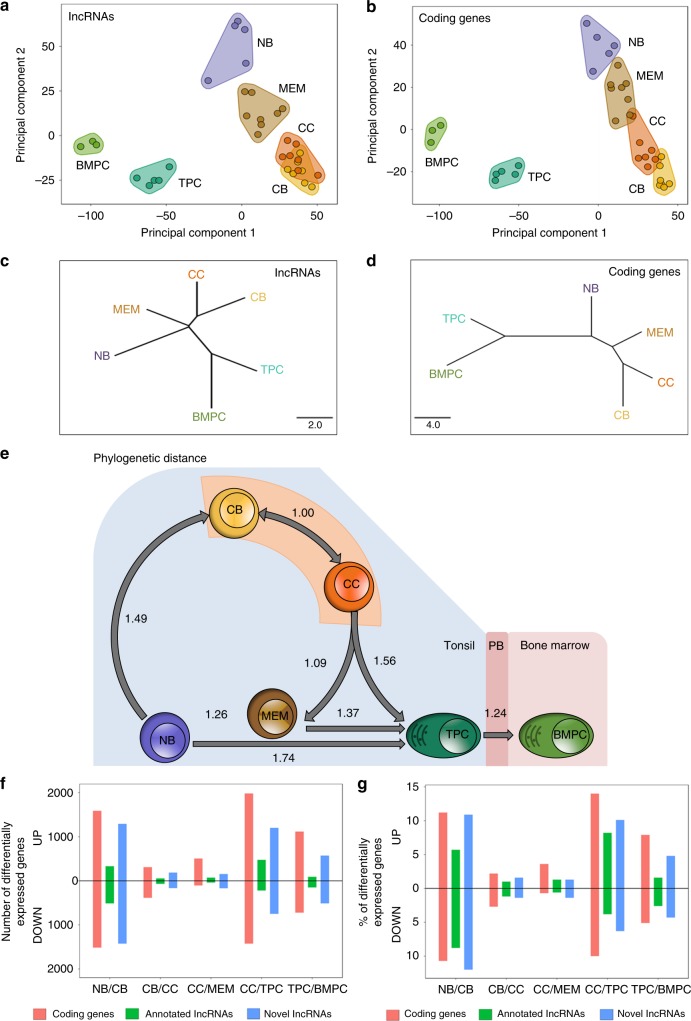
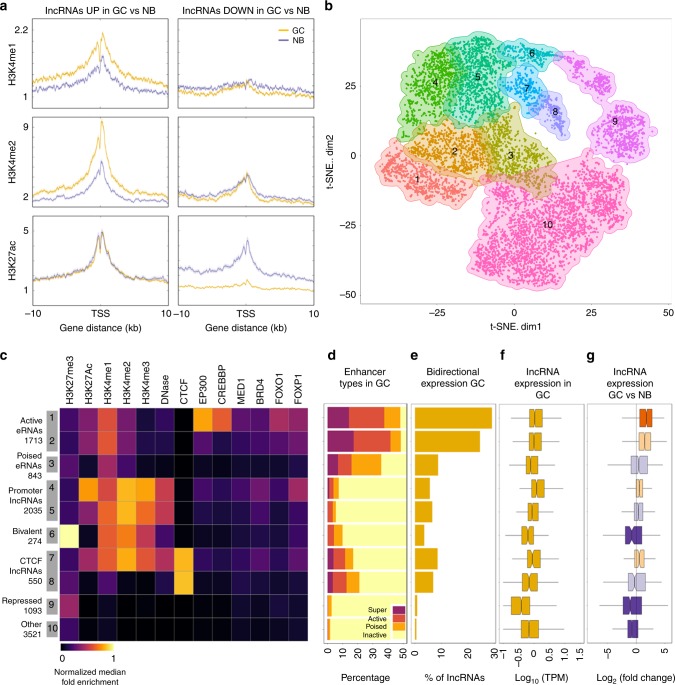
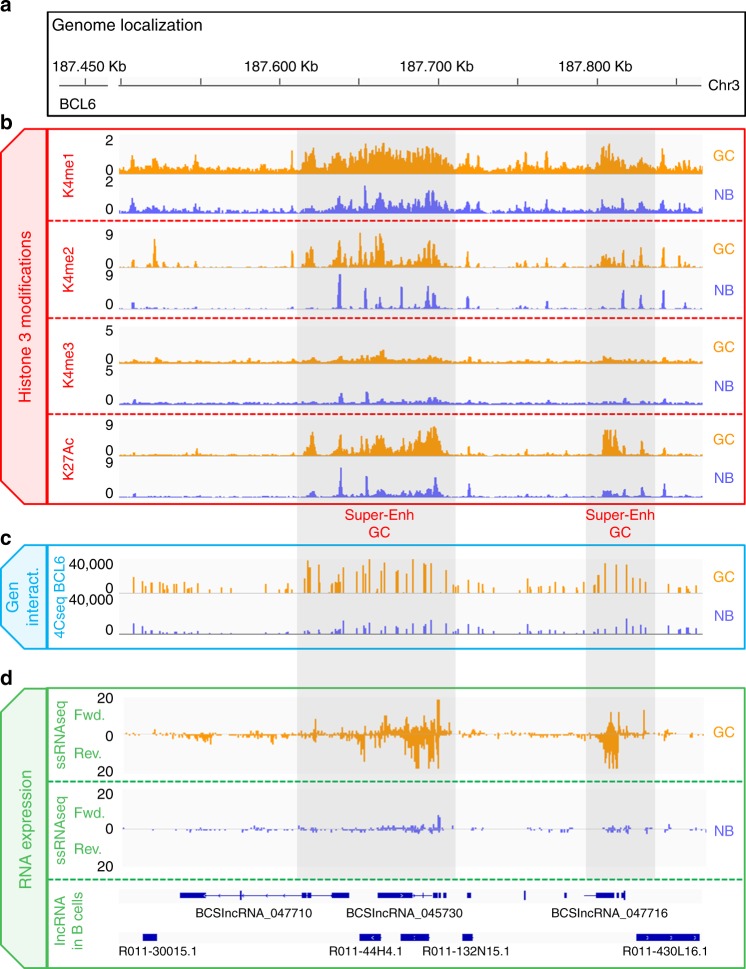
lncRNAs make up a majority of the human transcriptome and have key regulatory functions. Here we perform unbiased de novo annotation of transcripts expressed during the human humoral immune response to find 30% of the human genome transcribed during this process, yet 58% of these transcripts manifest striking differential expression, indicating an lncRNA phylogenetic relationship among cell types that is more robust than that of coding genes. We provide an atlas of lncRNAs in naive and GC B-cells that indicates their partition into ten functionally categories based on chromatin features, DNase hypersensitivity and transcription factor localization, defining lncRNAs classes such as enhancer-RNAs (eRNA), bivalent-lncRNAs, and CTCF-associated, among others. Specifically, eRNAs are transcribed in 8.6% of regular enhancers and 36.5% of super enhancers, and are associated with coding genes that participate in critical immune regulatory pathways, while plasma cells have uniquely high levels of circular-RNAs accounted for by and reflecting the combinatorial clonal state of the Immunoglobulin loci.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
