

At the intersection of DNA damage and immune responses
- PMID: 30778174
- PMCID: PMC6438741
- DOI: 10.1038/s41577-019-0135-6
At the intersection of DNA damage and immune responses
Abstract
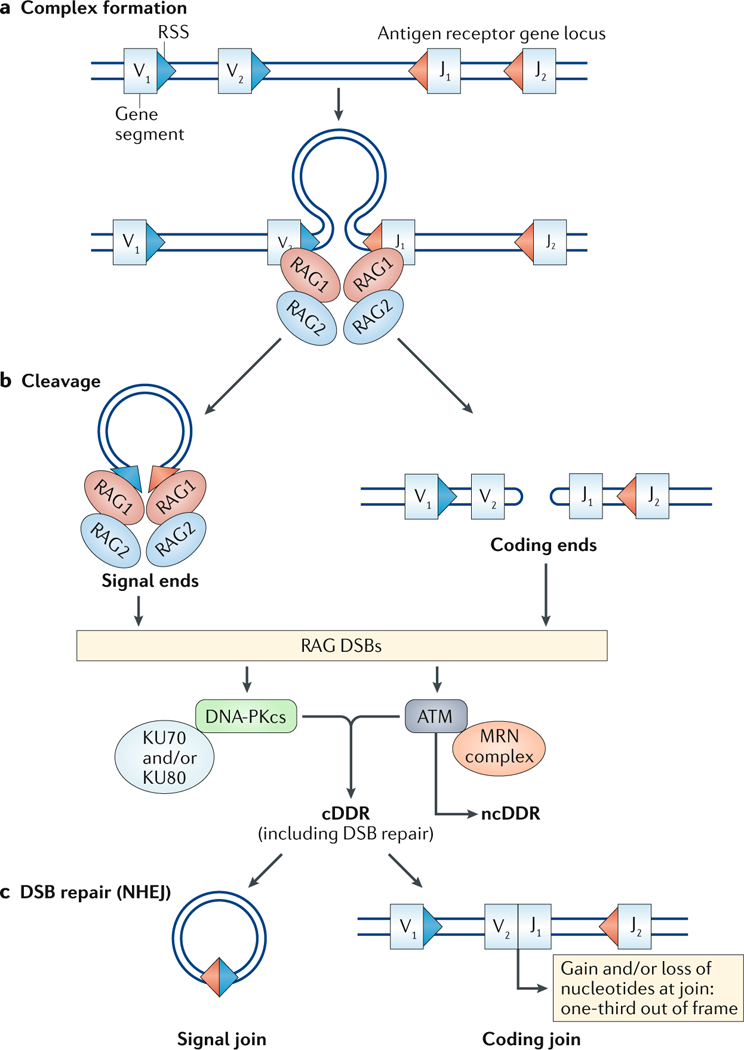
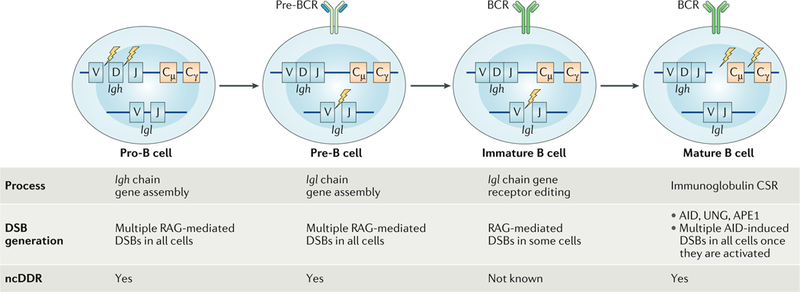
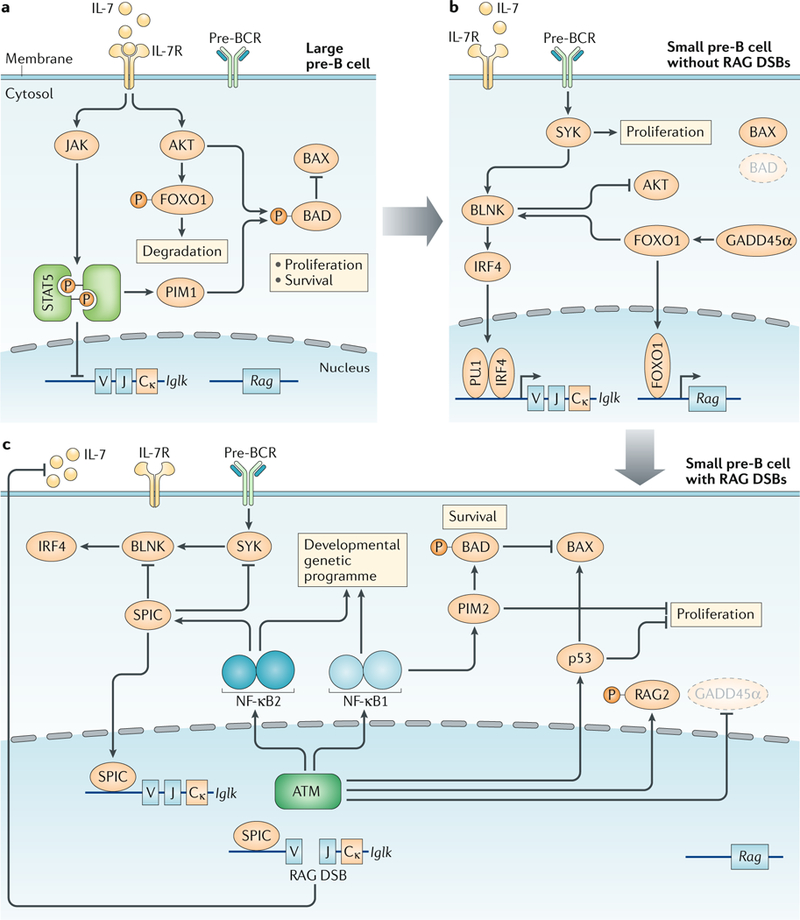
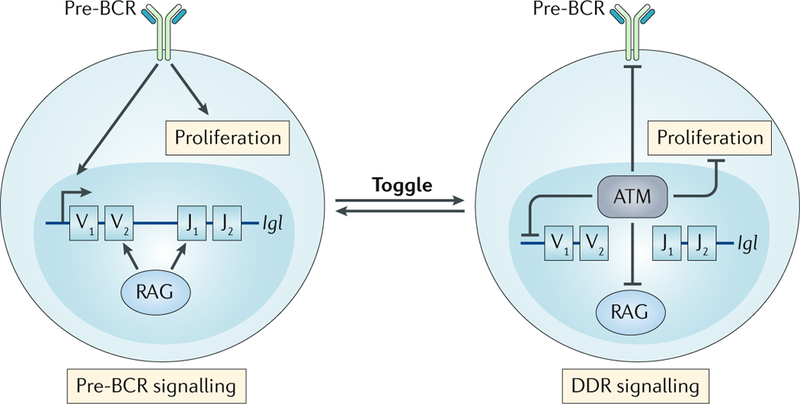
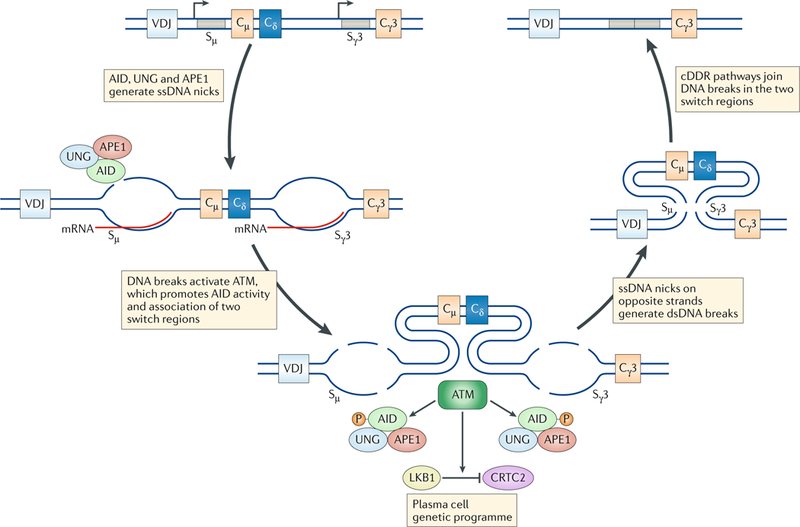
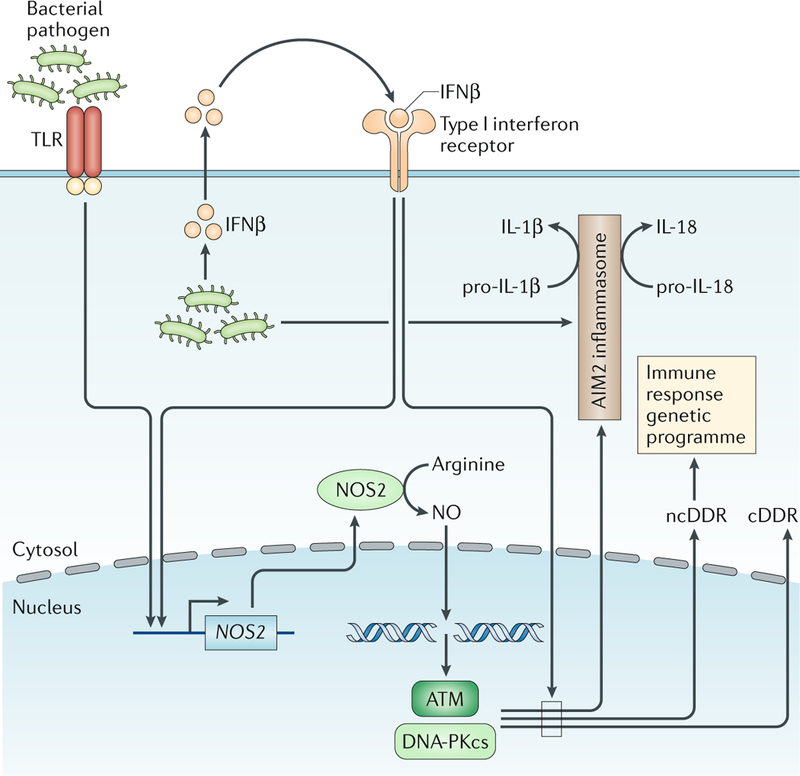
DNA damage occurs on exposure to genotoxic agents and during physiological DNA transactions. DNA double-strand breaks (DSBs) are particularly dangerous lesions that activate DNA damage response (DDR) kinases, leading to initiation of a canonical DDR (cDDR). This response includes activation of cell cycle checkpoints and engagement of pathways that repair the DNA DSBs to maintain genomic integrity. In adaptive immune cells, programmed DNA DSBs are generated at precise genomic locations during the assembly and diversification of lymphocyte antigen receptor genes. In innate immune cells, the production of genotoxic agents, such as reactive nitrogen molecules, in response to pathogens can also cause genomic DNA DSBs. These DSBs in adaptive and innate immune cells activate the cDDR. However, recent studies have demonstrated that they also activate non-canonical DDRs (ncDDRs) that regulate cell type-specific processes that are important for innate and adaptive immune responses. Here, we review these ncDDRs and discuss how they integrate with other signals during immune system development and function.
Figures

References
-
- Fugmann SD, Lee AI, Shockett PE, Villey IJ & Schatz DG The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu. Rev. Immunol. 18, 495–527 (2000). - PubMed
-
- Chaudhuri J et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv. Immunol. 94, 157–214 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
