

Multiplex chromatin interactions with single-molecule precision
- PMID: 30778195
- PMCID: PMC7001875
- DOI: 10.1038/s41586-019-0949-1
Multiplex chromatin interactions with single-molecule precision
Abstract
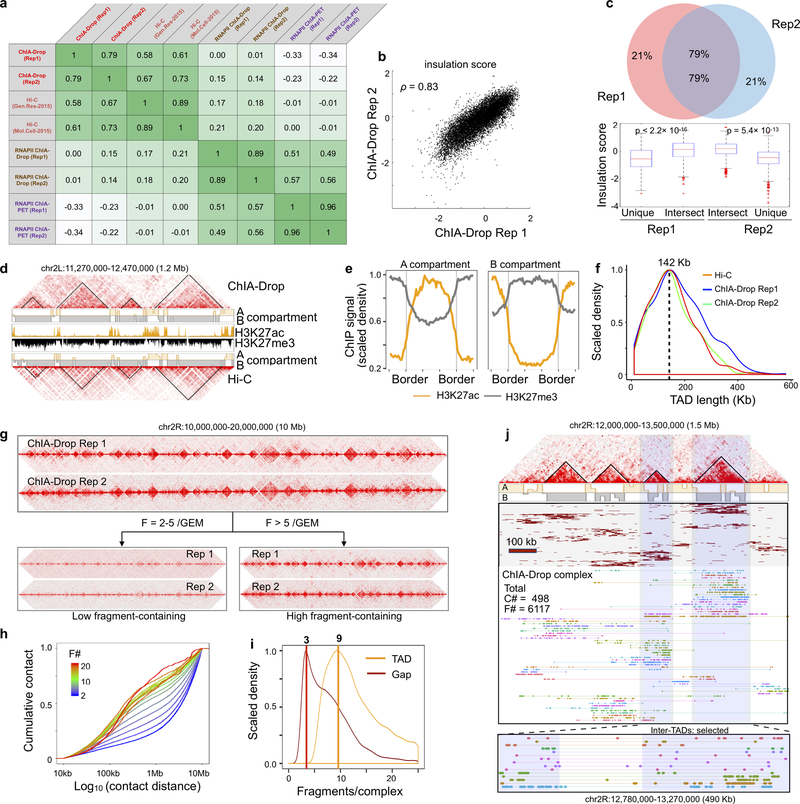
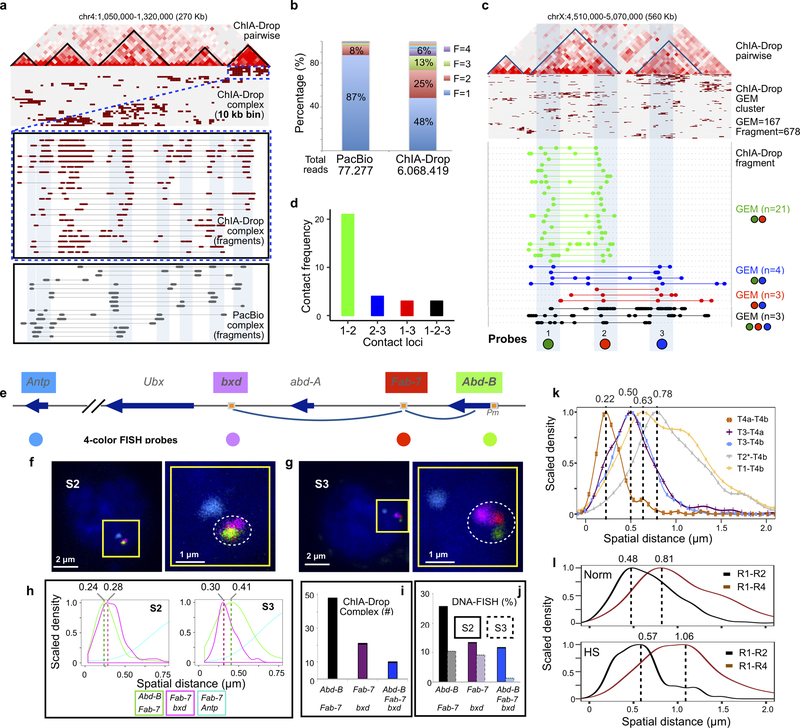
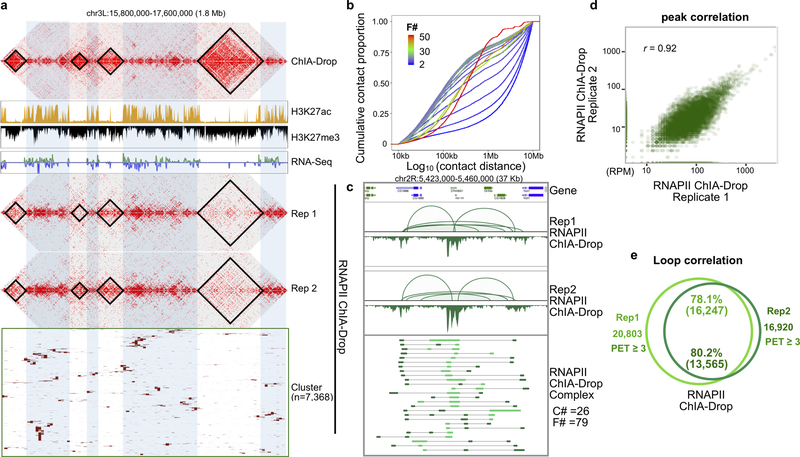
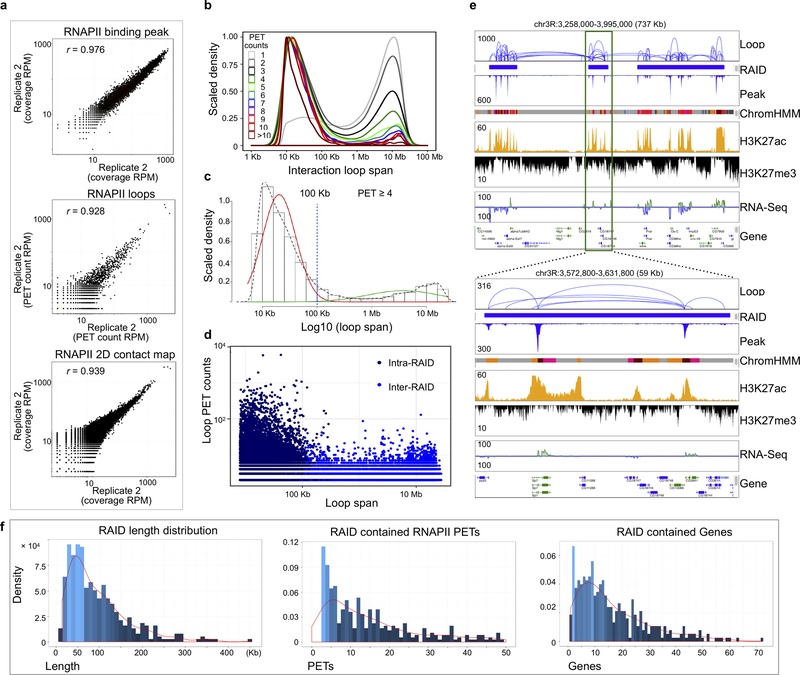
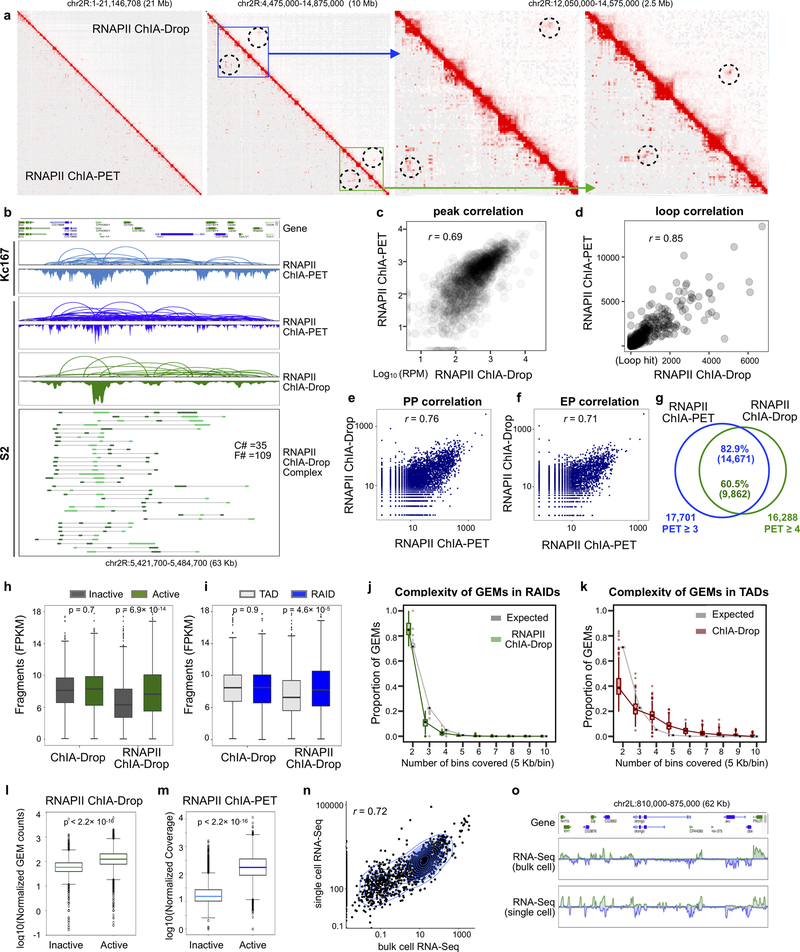
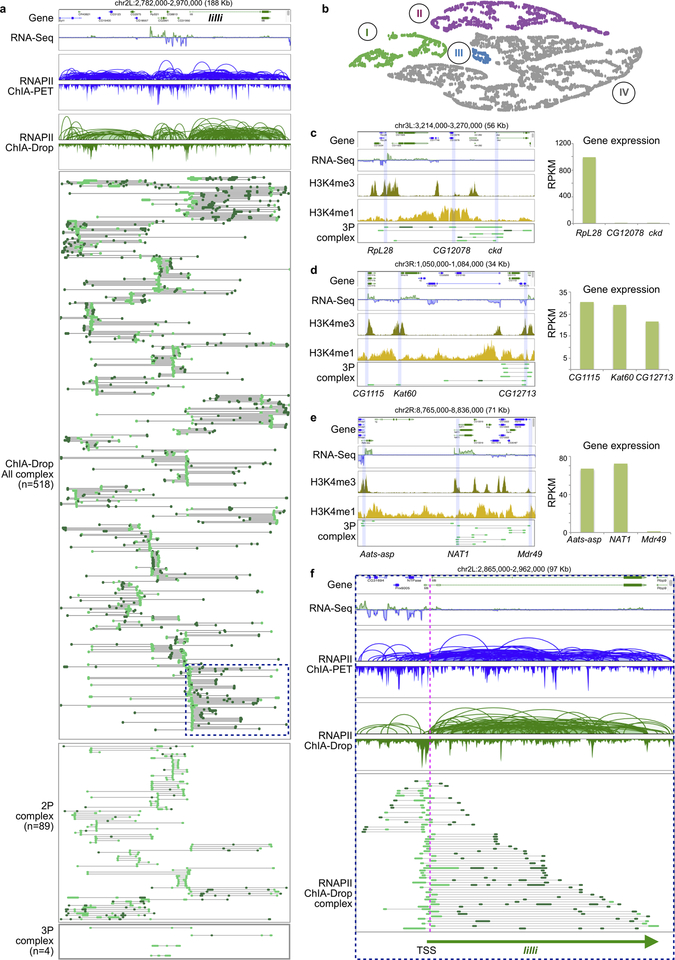
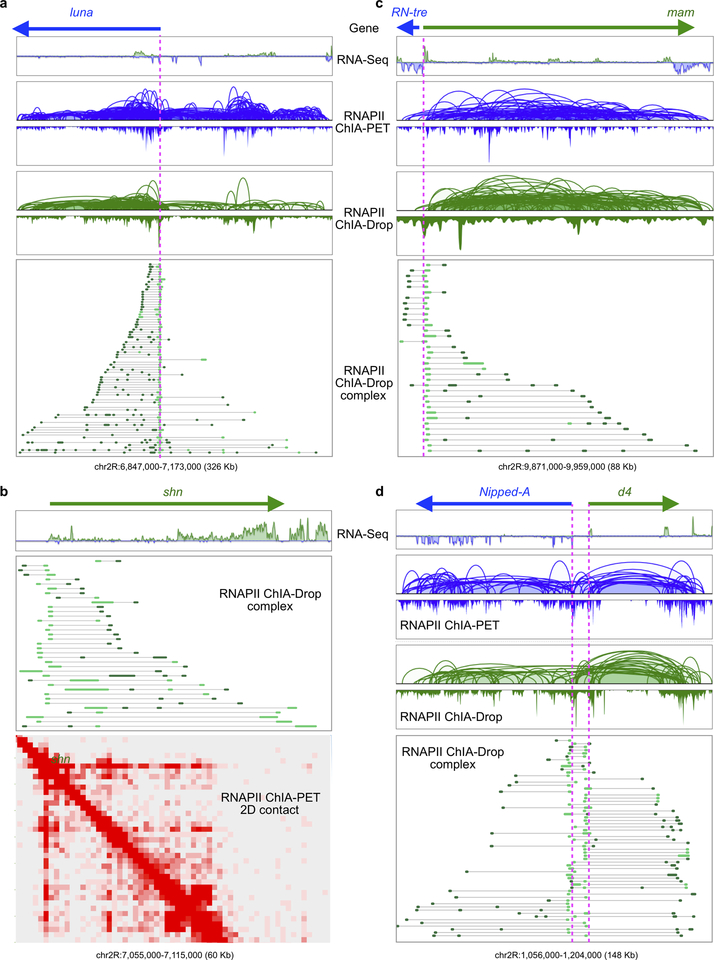
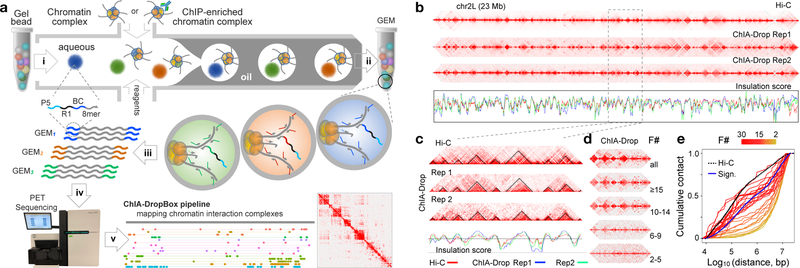
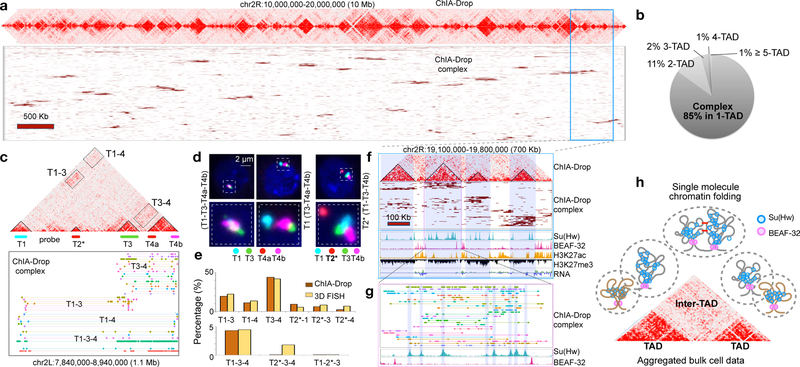
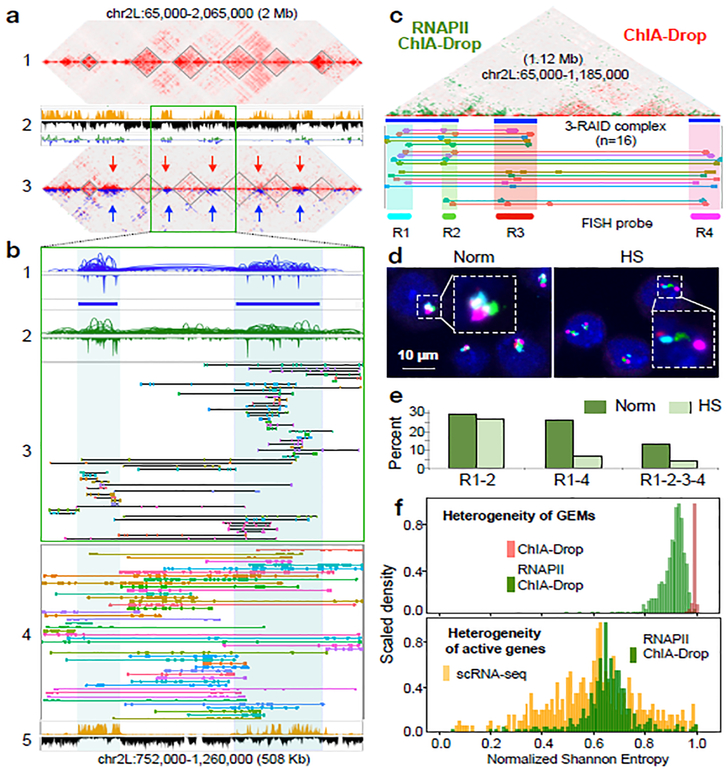
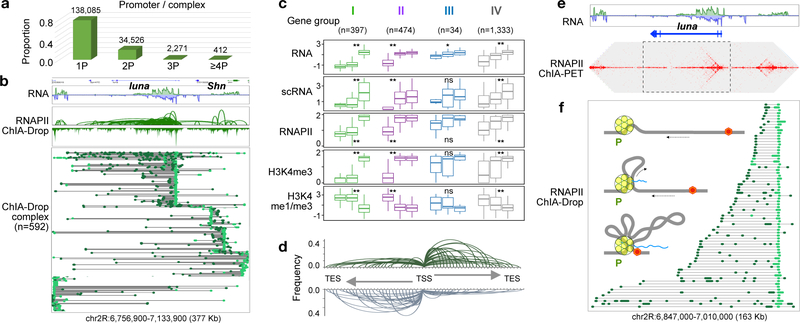
The genomes of multicellular organisms are extensively folded into 3D chromosome territories within the nucleus1. Advanced 3D genome-mapping methods that combine proximity ligation and high-throughput sequencing (such as chromosome conformation capture, Hi-C)2, and chromatin immunoprecipitation techniques (such as chromatin interaction analysis by paired-end tag sequencing, ChIA-PET)3, have revealed topologically associating domains4 with frequent chromatin contacts, and have identified chromatin loops mediated by specific protein factors for insulation and regulation of transcription5-7. However, these methods rely on pairwise proximity ligation and reflect population-level views, and thus cannot reveal the detailed nature of chromatin interactions. Although single-cell Hi-C8 potentially overcomes this issue, this method may be limited by the sparsity of data that is inherent to current single-cell assays. Recent advances in microfluidics have opened opportunities for droplet-based genomic analysis9 but this approach has not yet been adapted for chromatin interaction analysis. Here we describe a strategy for multiplex chromatin-interaction analysis via droplet-based and barcode-linked sequencing, which we name ChIA-Drop. We demonstrate the robustness of ChIA-Drop in capturing complex chromatin interactions with single-molecule precision, which has not been possible using methods based on population-level pairwise contacts. By applying ChIA-Drop to Drosophila cells, we show that chromatin topological structures predominantly consist of multiplex chromatin interactions with high heterogeneity; ChIA-Drop also reveals promoter-centred multivalent interactions, which provide topological insights into transcription.
Conflict of interest statement
The authors declare no competing interests.
Figures

Comment in
-
Getting the drop on chromatin interaction.Nat Rev Genet. 2019 Apr;20(4):192-193. doi: 10.1038/s41576-019-0103-9. Nat Rev Genet. 2019. PMID: 30783222 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
