

Astroglia in Sepsis Associated Encephalopathy
- PMID: 30778837
- PMCID: PMC7089215
- DOI: 10.1007/s11064-019-02743-2
Astroglia in Sepsis Associated Encephalopathy
Abstract
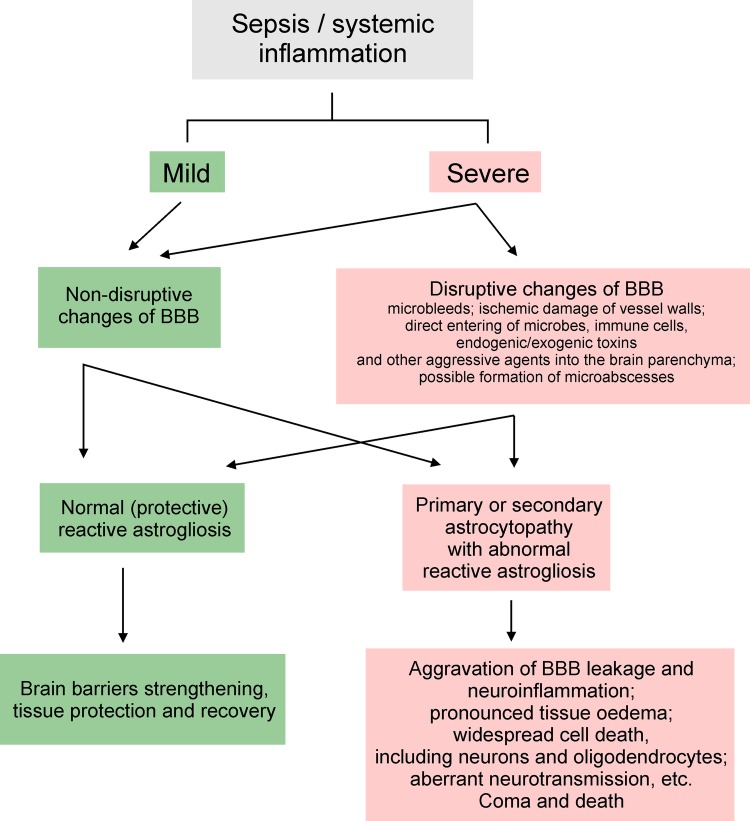
Cellular pathophysiology of sepsis associated encephalopathy (SAE) remains poorly characterised. Brain pathology in SAE, which is manifested by impaired perception, consciousness and cognition, results from multifactorial events, including high levels of systemic cytokines, microbial components and endotoxins, which all damage the brain barriers, instigate neuroinflammation and cause homeostatic failure. Astrocytes, being the principal homeostatic cells of the central nervous system contribute to the brain defence against infection. Forming multifunctional anatomical barriers, astroglial cells maintain brain-systemic interfaces and restrict the damage to the nervous tissue. Astrocytes detect, produce and integrate inflammatory signals between immune cells and cells of brain parenchyma, thus regulating brain immune response. In SAE astrocytes are present in both reactive and astrogliopathic states; balance between these states define evolution of pathology and neurological outcomes. In humans pathophysiology of SAE is complicated by frequent presence of comorbidities, as well as age-related remodelling of the brain tissue with senescence of astroglia; these confounding factors further impact upon SAE progression and neurological deficits.
Keywords: Asrtogliopathy; Astrocyte reactivity; Astroglia; Blood brain barrier; Infection; Sepsis associated encephalopathy; Sepsis signalling.
Figures



Similar articles
-
Sepsis-Associated Encephalopathy and Blood-Brain Barrier Dysfunction.Inflammation. 2021 Dec;44(6):2143-2150. doi: 10.1007/s10753-021-01501-3. Epub 2021 Jul 21. Inflammation. 2021. PMID: 34291398 Free PMC article. Review.
-
Mobilisation and redistribution of multivesicular bodies to the endfeet of reactive astrocytes in acute endogenous toxic encephalopathies.Brain Res. 2021 Jan 15;1751:147174. doi: 10.1016/j.brainres.2020.147174. Epub 2020 Oct 24. Brain Res. 2021. PMID: 33172595
-
Adenosine triggers early astrocyte reactivity that provokes microglial responses and drives the pathogenesis of sepsis-associated encephalopathy in mice.Nat Commun. 2024 Jul 27;15(1):6340. doi: 10.1038/s41467-024-50466-y. Nat Commun. 2024. PMID: 39068155 Free PMC article.
-
Sepsis-Associated Encephalopathy: from Pathophysiology to Progress in Experimental Studies.Mol Neurobiol. 2021 Jun;58(6):2770-2779. doi: 10.1007/s12035-021-02303-2. Epub 2021 Jan 26. Mol Neurobiol. 2021. PMID: 33495934 Review.
-
[Research progress on the changes of blood-brain barrier in sepsis-associated encephalopathy].Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2024 Aug;36(8):892-896. doi: 10.3760/cma.j.cn121430-20231109-00959. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2024. PMID: 39238417 Review. Chinese.
Cited by
-
Sepsis-Associated Encephalopathy: From Delirium to Dementia?J Clin Med. 2020 Mar 5;9(3):703. doi: 10.3390/jcm9030703. J Clin Med. 2020. PMID: 32150970 Free PMC article. Review.
-
NU9056, a KAT 5 Inhibitor, Treatment Alleviates Brain Dysfunction by Inhibiting NLRP3 Inflammasome Activation, Affecting Gut Microbiota, and Derived Metabolites in LPS-Treated Mice.Front Nutr. 2021 Jul 13;8:701760. doi: 10.3389/fnut.2021.701760. eCollection 2021. Front Nutr. 2021. PMID: 34327209 Free PMC article.
-
Gut Microbiota Regulate Astrocytic Functions in the Brain: Possible Therapeutic Consequences.Curr Neuropharmacol. 2021;19(8):1354-1366. doi: 10.2174/1570159X19666210215123239. Curr Neuropharmacol. 2021. PMID: 33588733 Free PMC article.
-
Combined cerebral oxygen saturation and neuron-specific enolase evaluation for diagnosis and prognosis of sepsis-associated encephalopathy.Sci Rep. 2025 May 2;15(1):15369. doi: 10.1038/s41598-025-00353-3. Sci Rep. 2025. PMID: 40316550 Free PMC article.
-
Convergence of sepsis-associated encephalopathy pathogenesis onto microglia.J Transl Med. 2025 Jun 3;23(1):622. doi: 10.1186/s12967-025-06635-8. J Transl Med. 2025. PMID: 40462103 Free PMC article. Review.
References
-
- Dal-Pizzol F, Tomasi CD, Ritter C. Septic encephalopathy: does inflammation drive the brain crazy? Rev Bras Psiquiatr. 2014;36:251–258. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
