

Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma
- PMID: 30782609
- PMCID: PMC6460420
- DOI: 10.1182/blood-2018-09-872549
Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma
Abstract
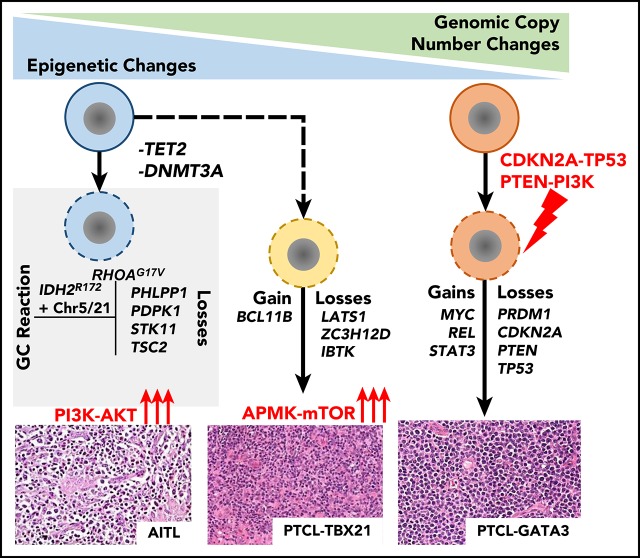
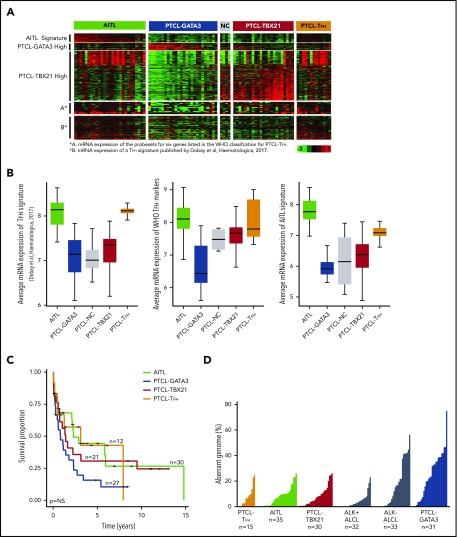
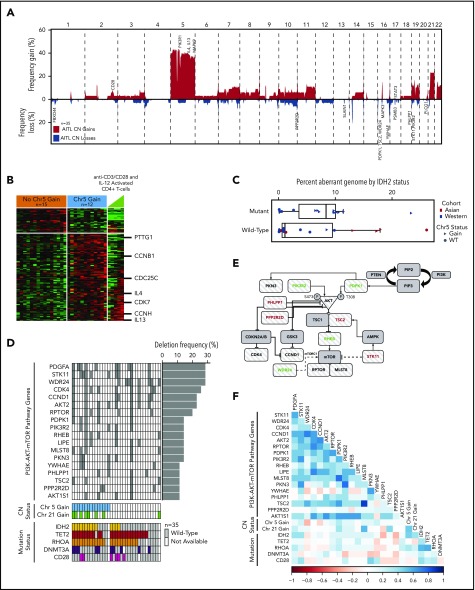
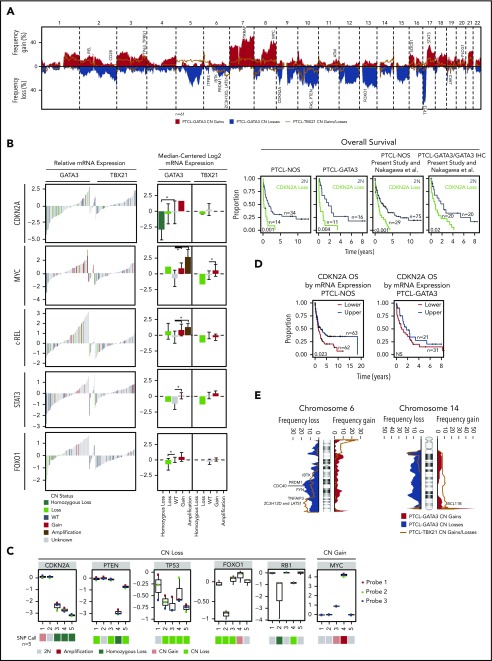
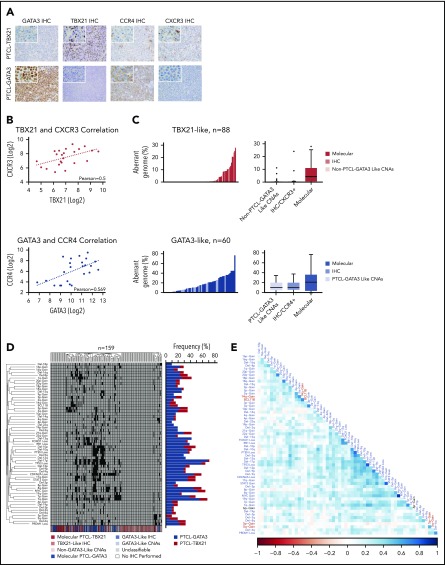
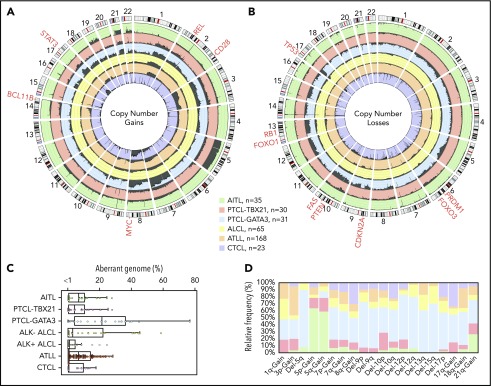
Peripheral T-cell lymphoma (PTCL) is a group of complex clinicopathological entities, often associated with an aggressive clinical course. Angioimmunoblastic T-cell lymphoma (AITL) and PTCL-not otherwise specified (PTCL-NOS) are the 2 most frequent categories, accounting for >50% of PTCLs. Gene expression profiling (GEP) defined molecular signatures for AITL and delineated biological and prognostic subgroups within PTCL-NOS (PTCL-GATA3 and PTCL-TBX21). Genomic copy number (CN) analysis and targeted sequencing of these molecular subgroups revealed unique CN abnormalities (CNAs) and oncogenic pathways, indicating distinct oncogenic evolution. PTCL-GATA3 exhibited greater genomic complexity that was characterized by frequent loss or mutation of tumor suppressor genes targeting the CDKN2A /B-TP53 axis and PTEN-PI3K pathways. Co-occurring gains/amplifications of STAT3 and MYC occurred in PTCL-GATA3. Several CNAs, in particular loss of CDKN2A, exhibited prognostic significance in PTCL-NOS as a single entity and in the PTCL-GATA3 subgroup. The PTCL-TBX21 subgroup had fewer CNAs, primarily targeting cytotoxic effector genes, and was enriched in mutations of genes regulating DNA methylation. CNAs affecting metabolic processes regulating RNA/protein degradation and T-cell receptor signaling were common in both subgroups. AITL showed lower genomic complexity compared with other PTCL entities, with frequent co-occurring gains of chromosome 5 (chr5) and chr21 that were significantly associated with IDH2 R172 mutation. CN losses were enriched in genes regulating PI3K-AKT-mTOR signaling in cases without IDH2 mutation. Overall, we demonstrated that novel GEP-defined PTCL subgroups likely evolve by distinct genetic pathways and provided biological rationale for therapies that may be investigated in future clinical trials.
© 2019 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures

References
-
- Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Vol. 2. Revised 4th ed. Lyon, France: International Agency for Cancer Research; 2017.
-
- Vose J, Armitage J, Weisenburger D; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124-4130. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
