

Synaptic retinoic acid receptor signaling mediates mTOR-dependent metaplasticity that controls hippocampal learning
- PMID: 30782829
- PMCID: PMC6452649
- DOI: 10.1073/pnas.1820690116
Synaptic retinoic acid receptor signaling mediates mTOR-dependent metaplasticity that controls hippocampal learning
Abstract
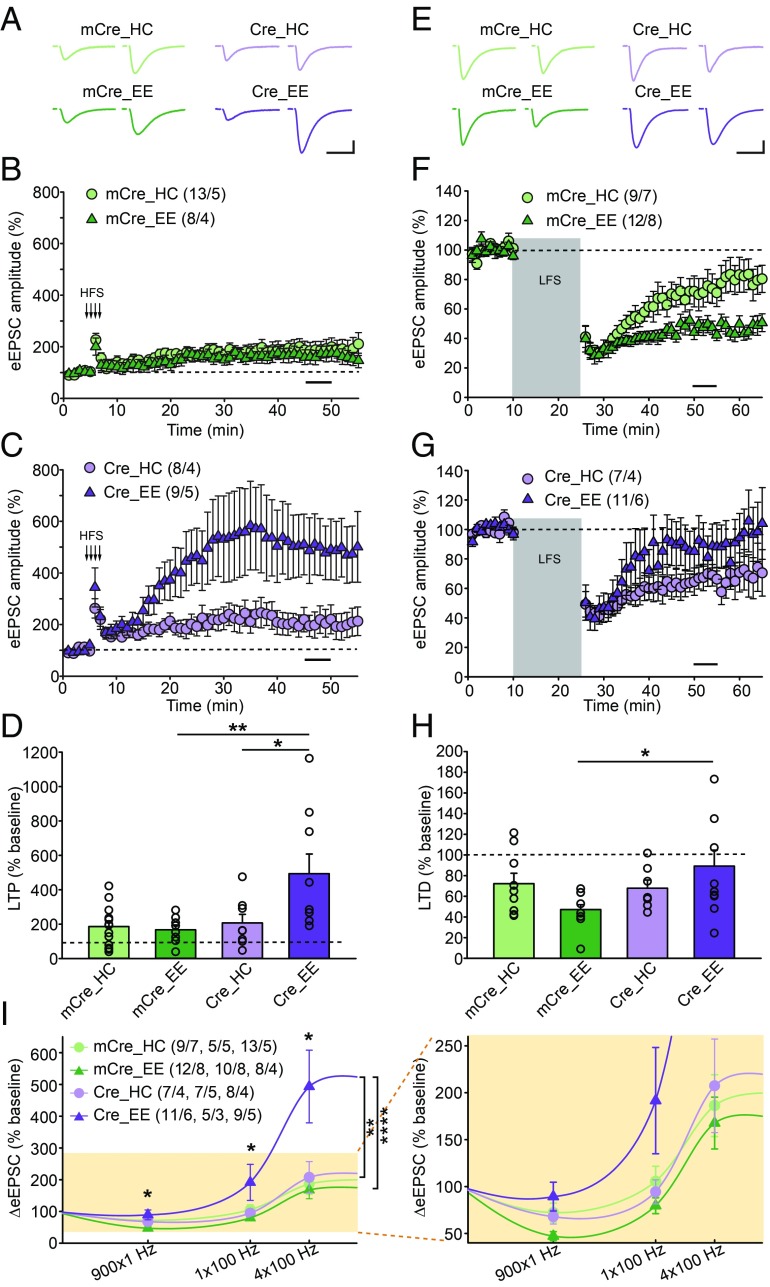
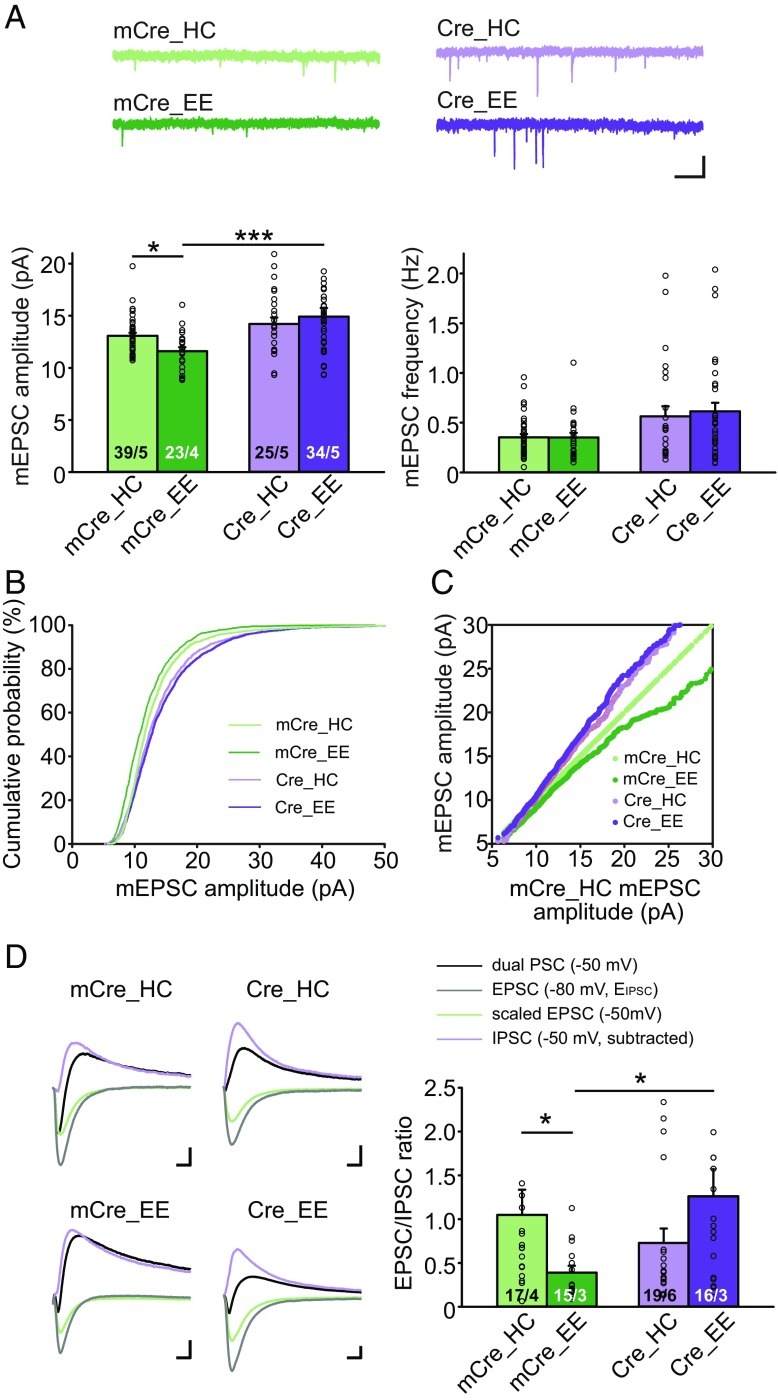
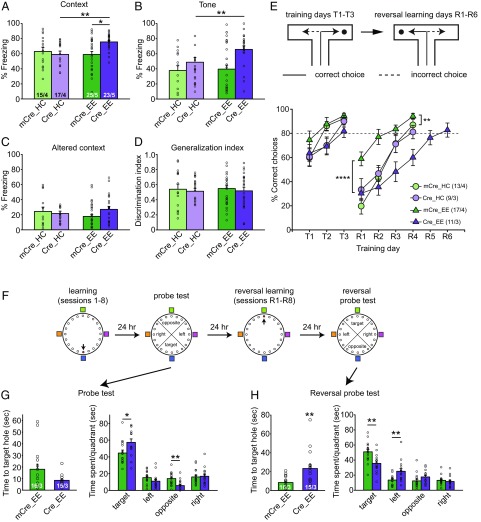
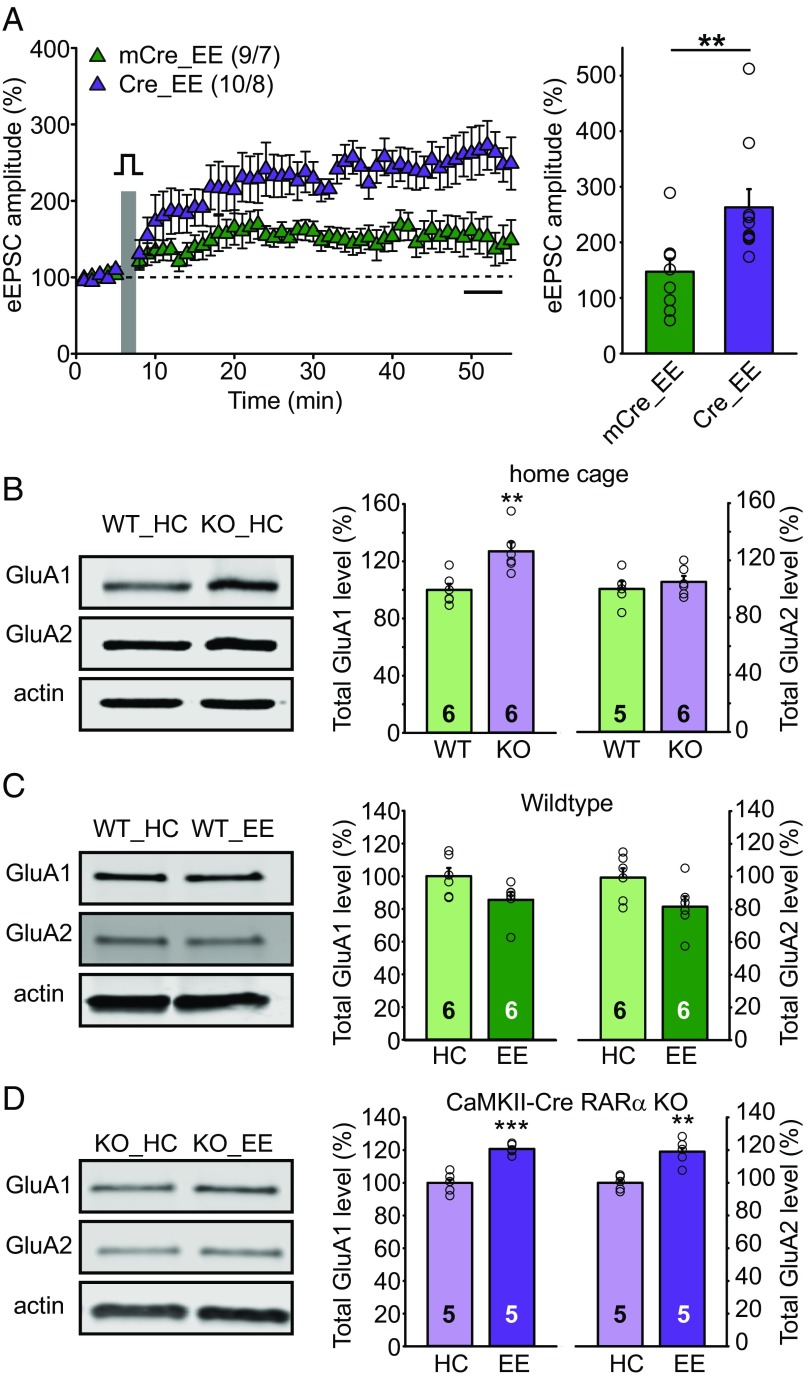
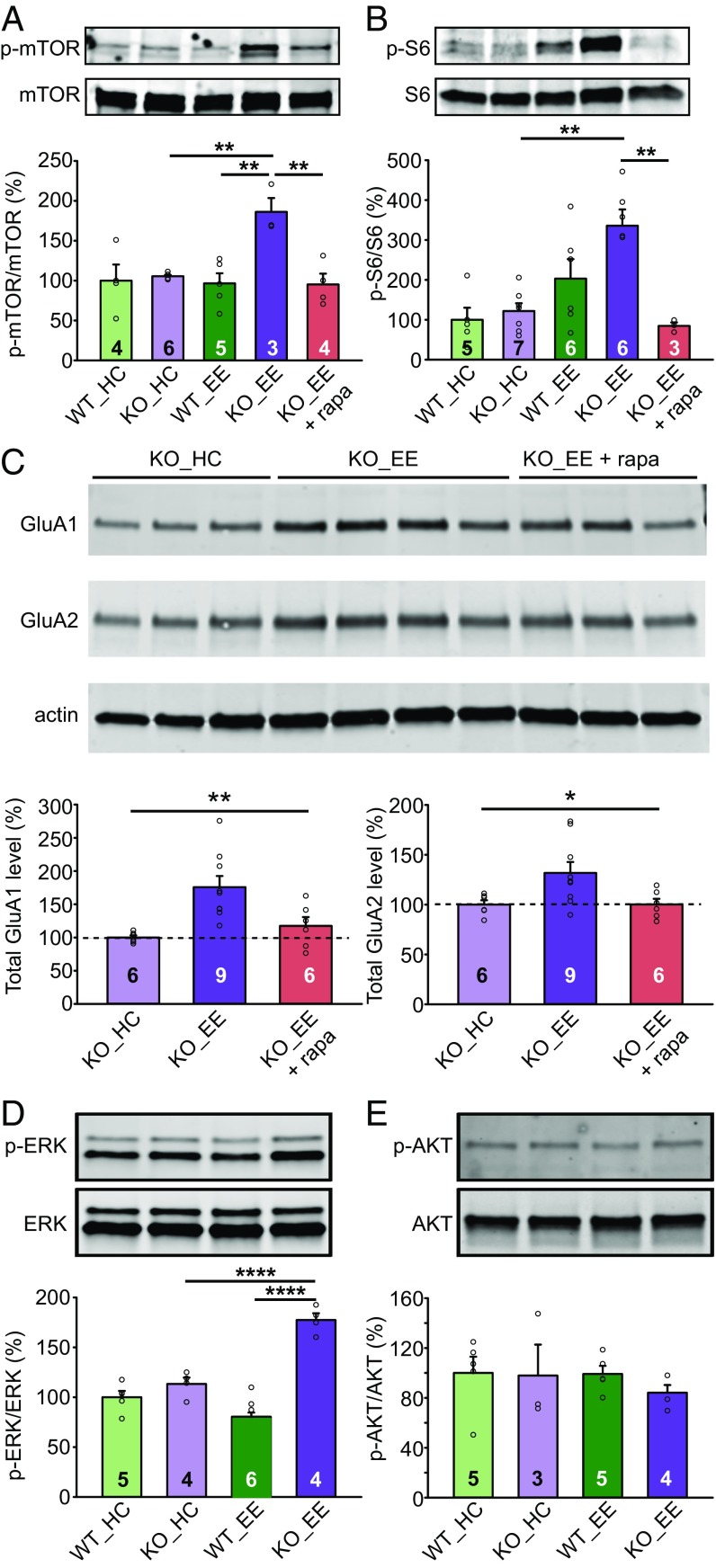
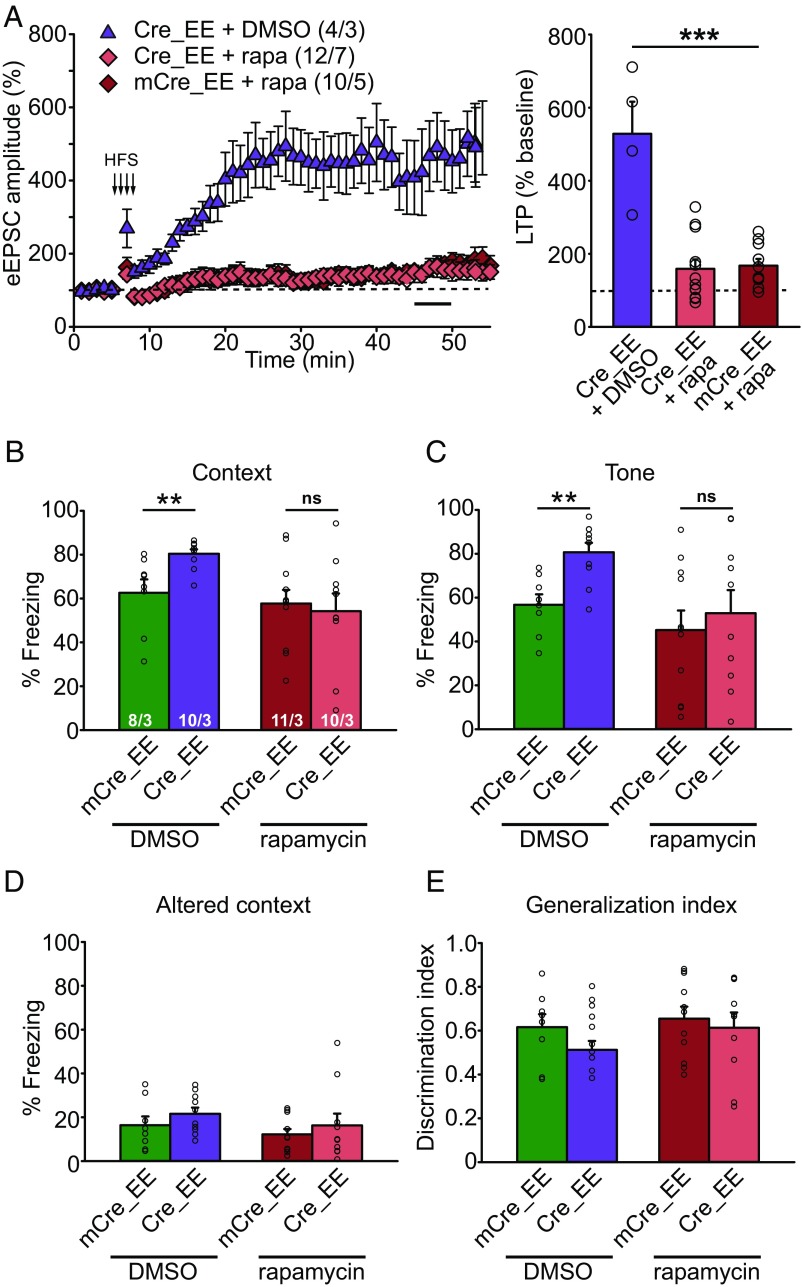
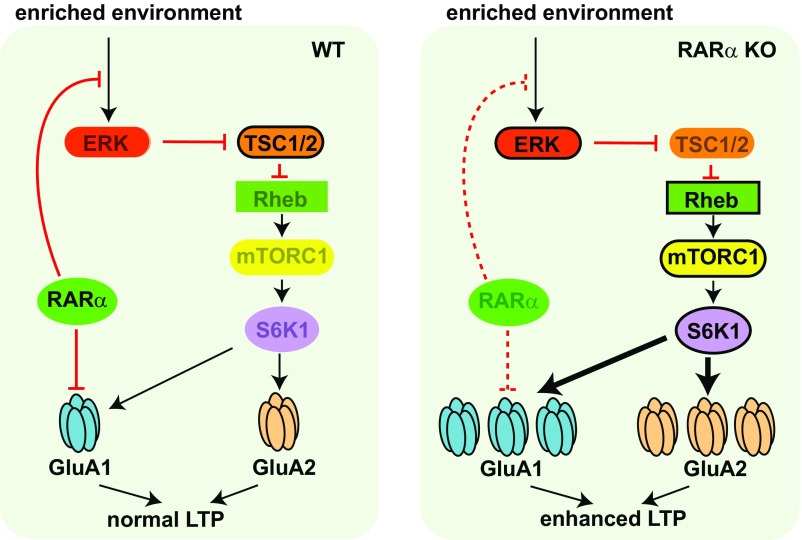
Homeostatic synaptic plasticity is a stabilizing mechanism engaged by neural circuits in response to prolonged perturbation of network activity. The non-Hebbian nature of homeostatic synaptic plasticity is thought to contribute to network stability by preventing "runaway" Hebbian plasticity at individual synapses. However, whether blocking homeostatic synaptic plasticity indeed induces runaway Hebbian plasticity in an intact neural circuit has not been explored. Furthermore, how compromised homeostatic synaptic plasticity impacts animal learning remains unclear. Here, we show in mice that the experience of an enriched environment (EE) engaged homeostatic synaptic plasticity in hippocampal circuits, thereby reducing excitatory synaptic transmission. This process required RARα, a nuclear retinoic acid receptor that doubles as a cytoplasmic retinoic acid-induced postsynaptic regulator of protein synthesis. Blocking RARα-dependent homeostatic synaptic plasticity during an EE experience by ablating RARα signaling induced runaway Hebbian plasticity, as evidenced by greatly enhanced long-term potentiation (LTP). As a consequence, RARα deletion in hippocampal circuits during an EE experience resulted in enhanced spatial learning but suppressed learning flexibility. In the absence of RARα, moreover, EE experience superactivated mammalian target of rapamycin (mTOR) signaling, causing a shift in protein translation that enhanced the expression levels of AMPA-type glutamate receptors. Treatment of mice with the mTOR inhibitor rapamycin during an EE experience not only restored normal AMPA-receptor expression levels but also reversed the increases in runaway Hebbian plasticity and learning after hippocampal RARα deletion. Thus, our findings reveal an RARα- and mTOR-dependent mechanism by which homeostatic plasticity controls Hebbian plasticity and learning.
Keywords: Hebbian plasticity; enriched environment; homeostatic synaptic plasticity; mTOR signaling; retinoic acid receptor.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Retinoic acid receptor plays both sides of homeostatic plasticity.Proc Natl Acad Sci U S A. 2019 Apr 2;116(14):6528-6530. doi: 10.1073/pnas.1902400116. Epub 2019 Mar 14. Proc Natl Acad Sci U S A. 2019. PMID: 30872478 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
