

Isoform-specific Ras signaling is growth factor dependent
- PMID: 30785867
- PMCID: PMC6724511
- DOI: 10.1091/mbc.E18-10-0676
Isoform-specific Ras signaling is growth factor dependent
Abstract
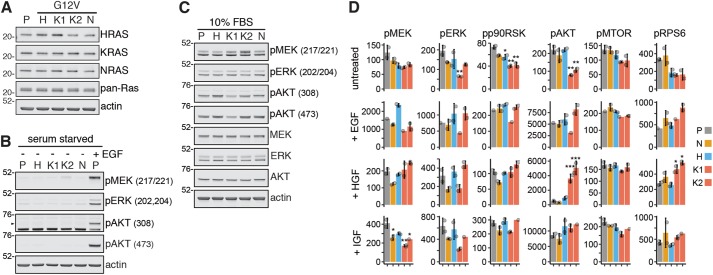
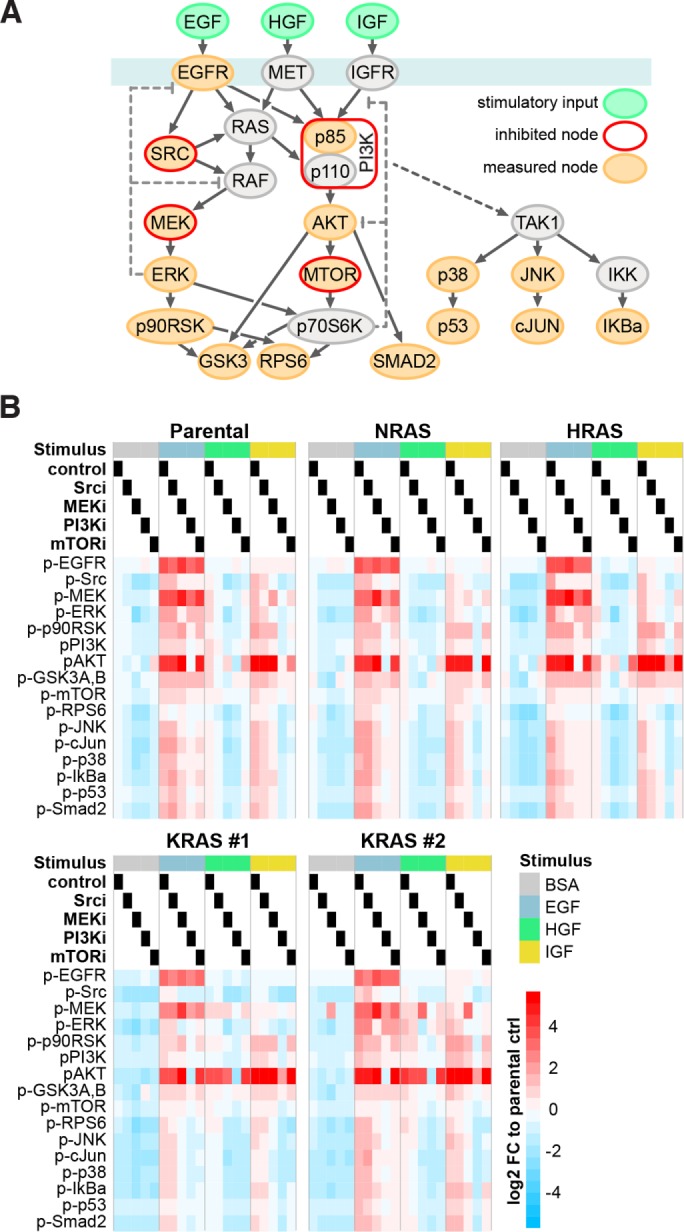
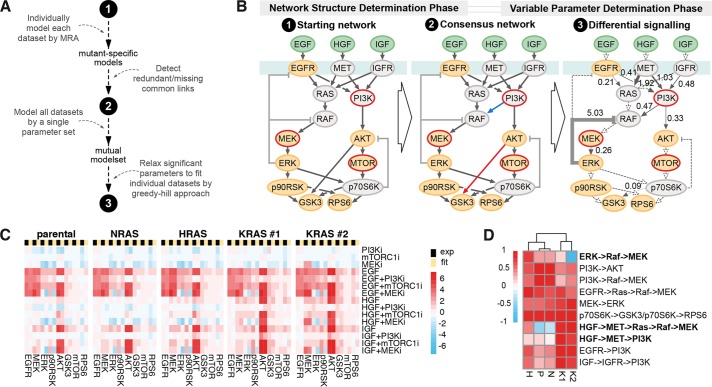
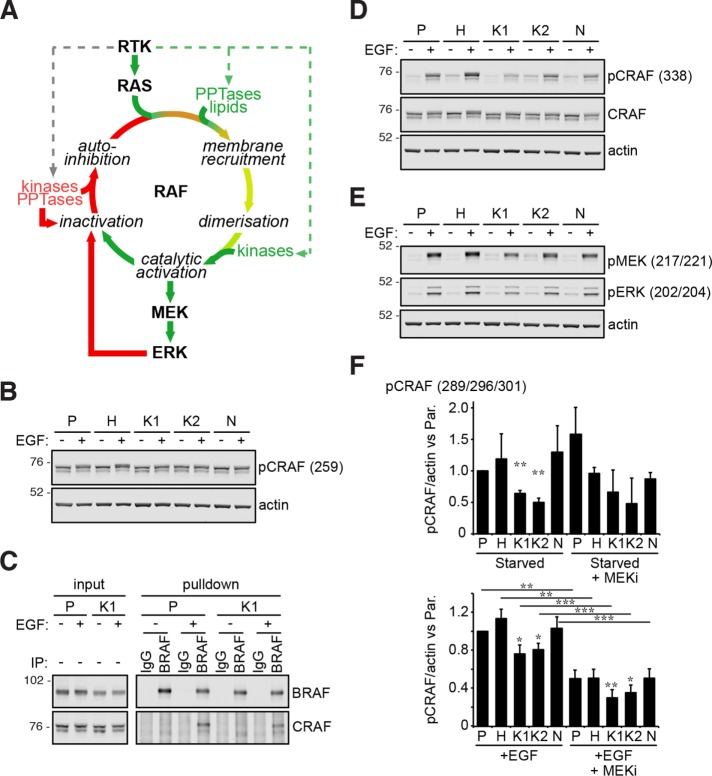
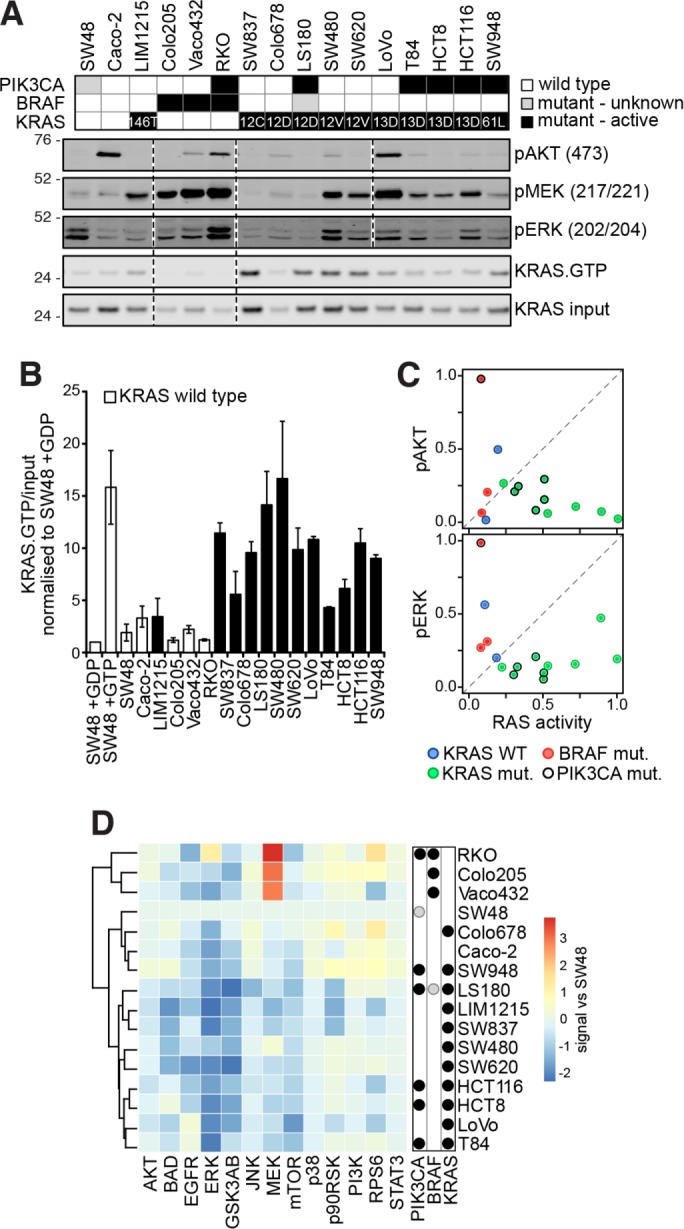
HRAS, NRAS, and KRAS isoforms are almost identical proteins that are ubiquitously expressed and activate a common set of effectors. In vivo studies have revealed that they are not biologically redundant; however, the isoform specificity of Ras signaling remains poorly understood. Using a novel panel of isogenic SW48 cell lines endogenously expressing wild-type or G12V-mutated activated Ras isoforms, we have performed a detailed characterization of endogenous isoform-specific mutant Ras signaling. We find that despite displaying significant Ras activation, the downstream outputs of oncogenic Ras mutants are minimal in the absence of growth factor inputs. The lack of mutant KRAS-induced effector activation observed in SW48 cells appears to be representative of a broad panel of colon cancer cell lines harboring mutant KRAS. For MAP kinase pathway activation in KRAS-mutant cells, the requirement for coincident growth factor stimulation occurs at an early point in the Raf activation cycle. Finally, we find that Ras isoform-specific signaling was highly context dependent and did not conform to the dogma derived from ectopic expression studies.
Figures
References
-
- Alamo P, Gallardo A, Di Nicolantonio F, Pavon MA, Casanova I, Trias M, Mangues MA, Lopez-Pousa A, Villaverde A, Vazquez E, et al (2015). Higher metastatic efficiency of KRas G12V than KRas G13D in a colorectal cancer model. FASEB J , 464–476. - PubMed
-
- De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, Lamba S, Arena S, Frattini M, Piessevaux H, et al (2010). Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. J Am Med Assoc , 1812–1820. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
