

The impact of Pompe disease on smooth muscle: a review
- PMID: 30787211
- PMCID: PMC6380904
- DOI: 10.1540/jsmr.54.100
The impact of Pompe disease on smooth muscle: a review
Abstract
Pompe disease (OMIM 232300) is an autosomal recessive disorder caused by mutations in the gene encoding acid α-glucosidase (GAA) (EC 3.2.1.20), the enzyme responsible for hydrolyzing lysosomal glycogen. The primary cellular pathology is lysosomal glycogen accumulation in cardiac muscle, skeletal muscle, and motor neurons, which ultimately results in cardiorespiratory failure. However, the severity of pathology and its impact on clinical outcomes are poorly described in smooth muscle. The advent of enzyme replacement therapy (ERT) in 2006 has improved clinical outcomes in infantile-onset Pompe disease patients. Although ERT increases patient life expectancy and ventilator free survival, it is not entirely curative. Persistent motor neuron pathology and weakness of respiratory muscles, including airway smooth muscles, contribute to the need for mechanical ventilation by some patients on ERT. Some patients on ERT continue to experience life-threatening pathology to vascular smooth muscle, such as aneurysms or dissections within the aorta and cerebral arteries. Better characterization of the disease impact on smooth muscle will inform treatment development and help anticipate later complications. This review summarizes the published knowledge of smooth muscle pathology associated with Pompe disease in animal models and in patients.
Keywords: Pompe disease; airway; gastrointestinal; smooth muscle; vasculature.
Figures
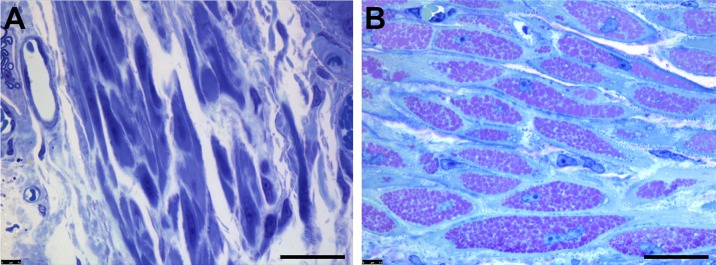
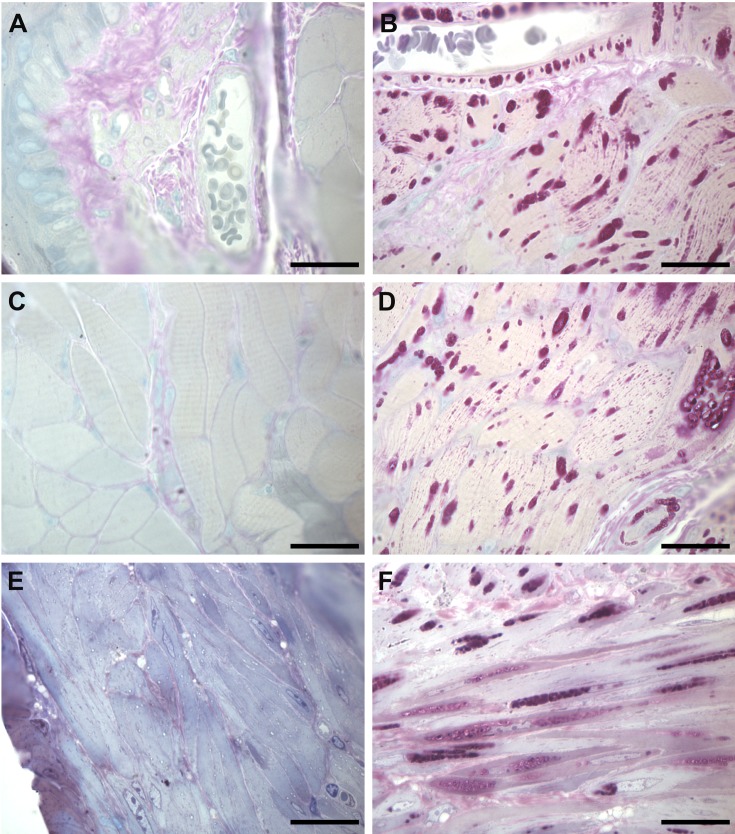
References
-
- van den Hout HMP, Hop W, van Diggelen OP, Smeitink JAM, Smit GPA, Poll-The BTT, Bakker HD, Loonen MCB, de Klerk JBC, Reuser AJJ, van der Ploeg AT. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003; 112(2): 332–40. doi: 10.1542/peds.112.2.332 - DOI - PubMed
-
- Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D, Infantile-Onset Pompe Disease Natural History Study Group. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006; 148(5): 671–6. doi: 10.1016/j.jpeds.2005.11.033 - DOI - PubMed
-
- Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, Crowley JF, Downs S, Howell RR, Kravitz RM, Mackey J, Marsden D, Martins AM, Millington DS, Nicolino M, O’Grady G, Patterson MC, Rapoport DM, Slonim A, Spencer CT, Tifft CJ, Watson MS. Pompe disease diagnosis and management guideline. Genet Med. 2006; 8(5): 267–88. doi: 10.1097/01.gim.0000218152.87434.f3 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous