

The single-cell transcriptional landscape of mammalian organogenesis
- PMID: 30787437
- PMCID: PMC6434952
- DOI: 10.1038/s41586-019-0969-x
The single-cell transcriptional landscape of mammalian organogenesis
Abstract
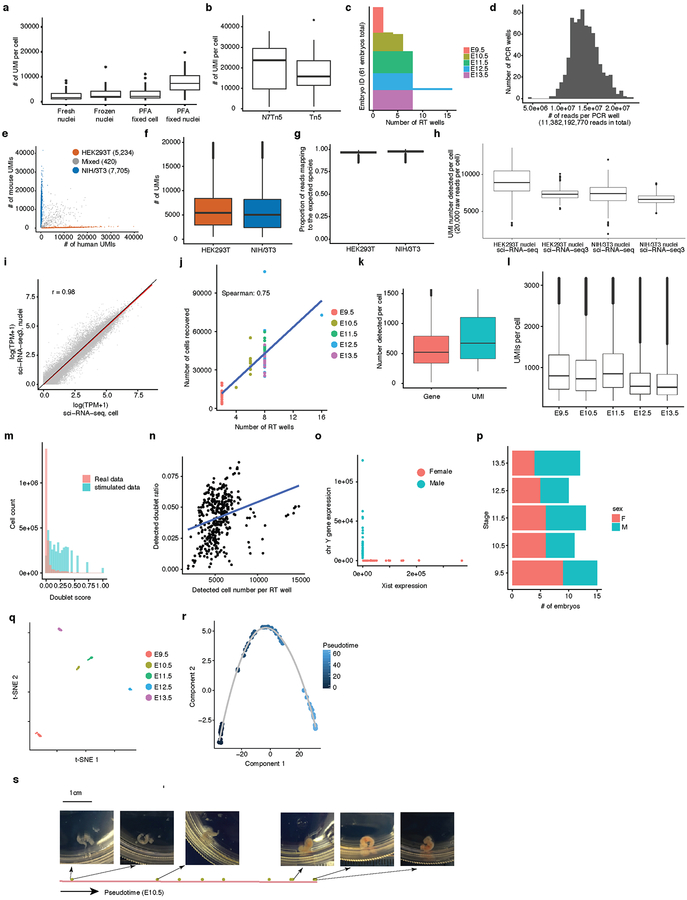
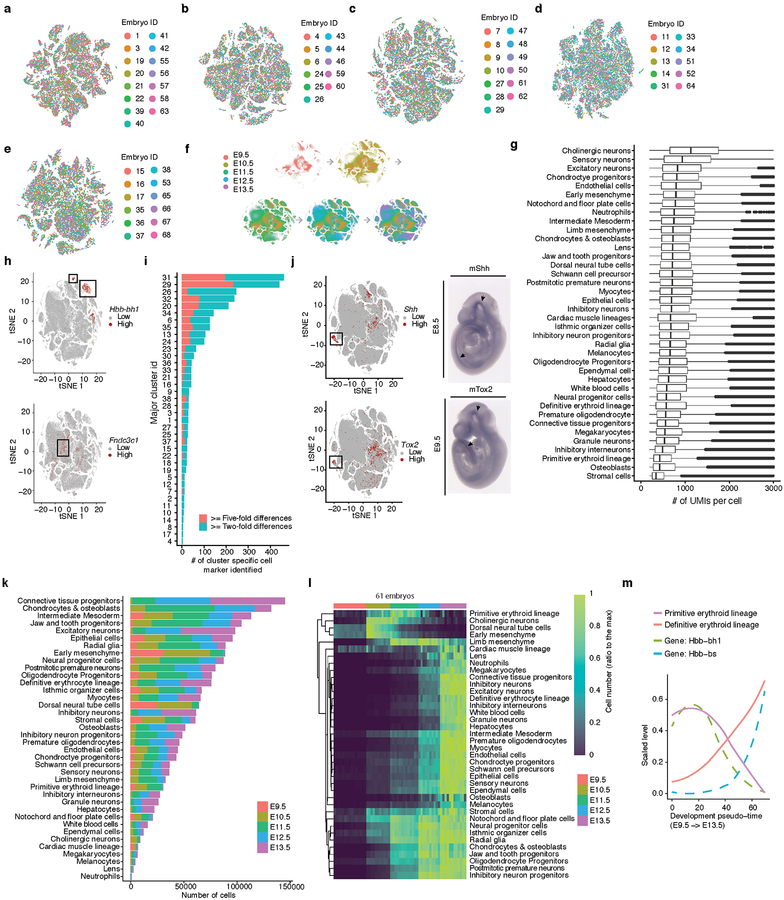
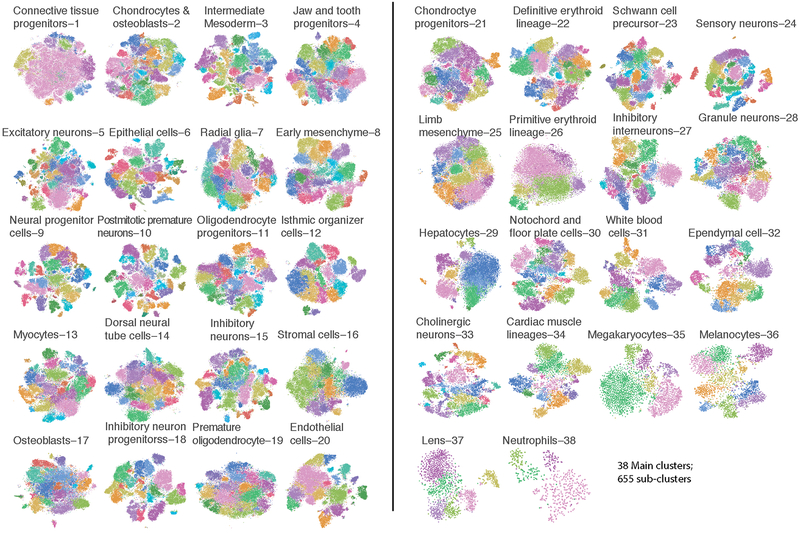
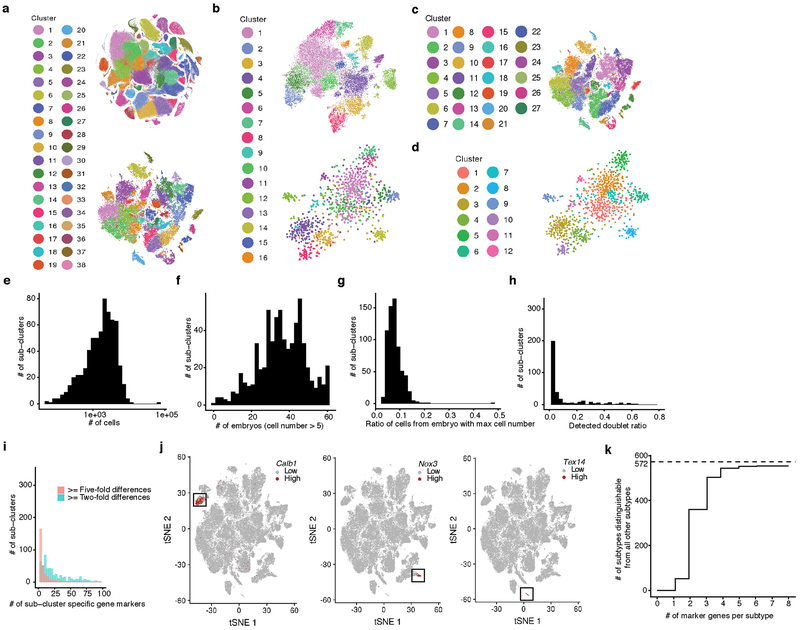
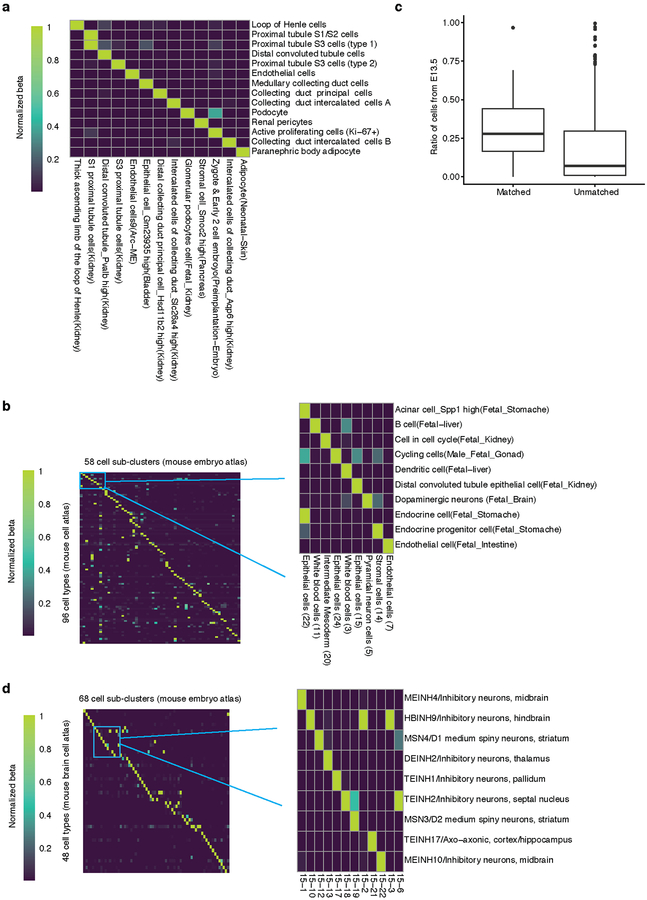
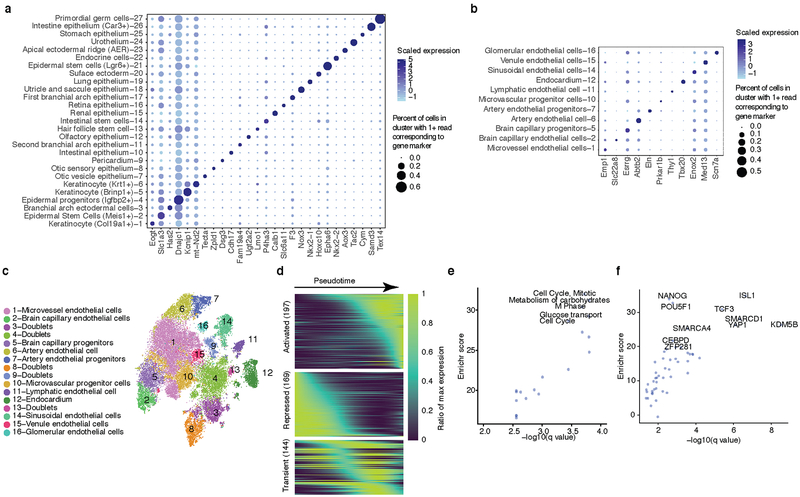
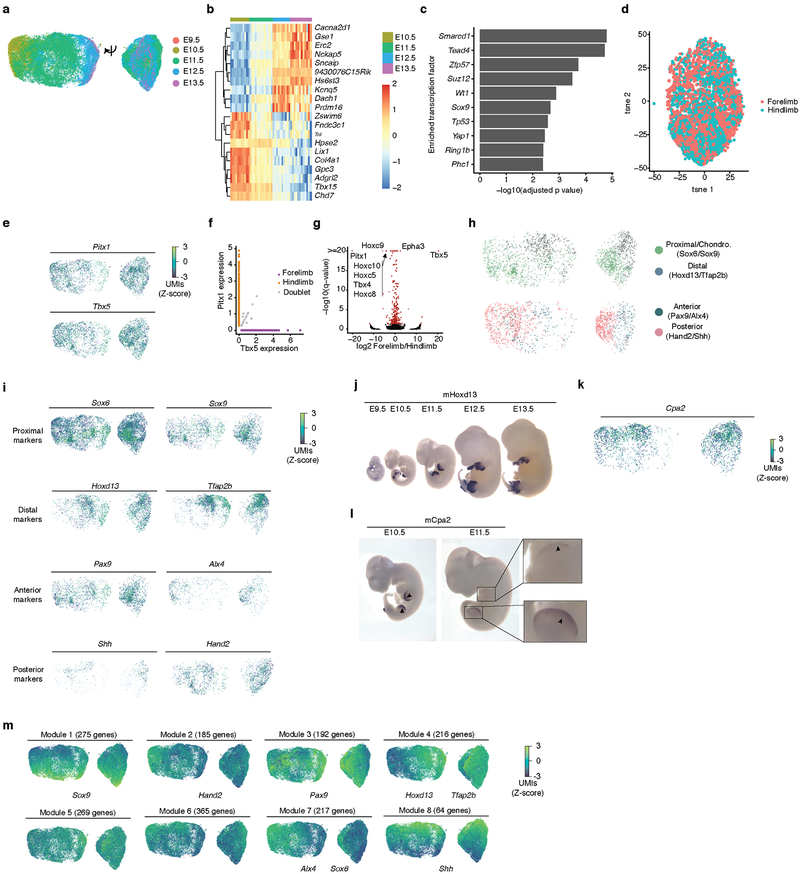
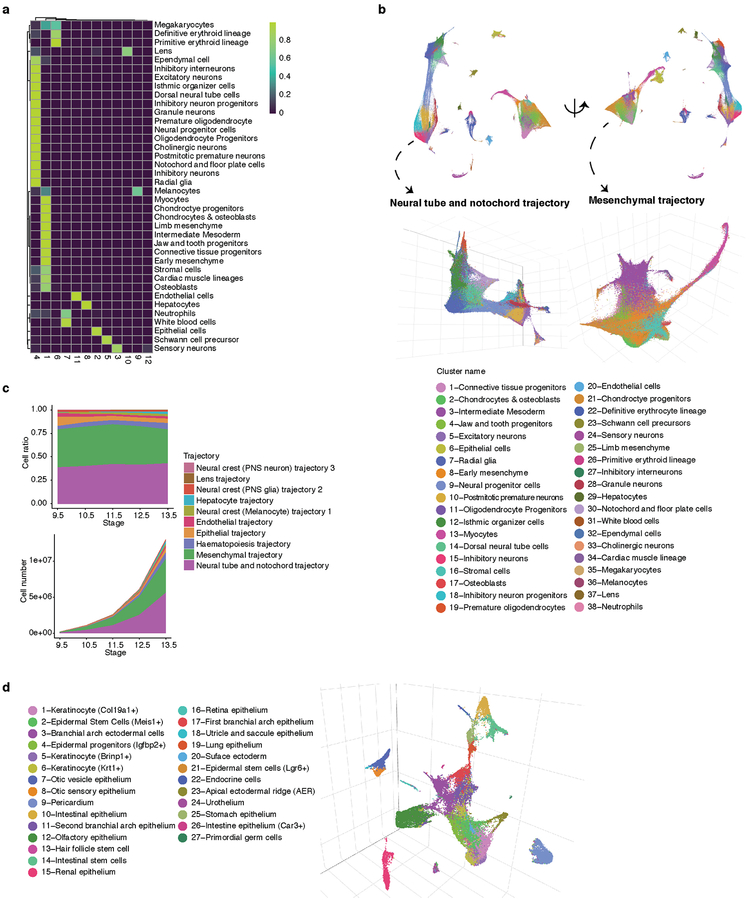
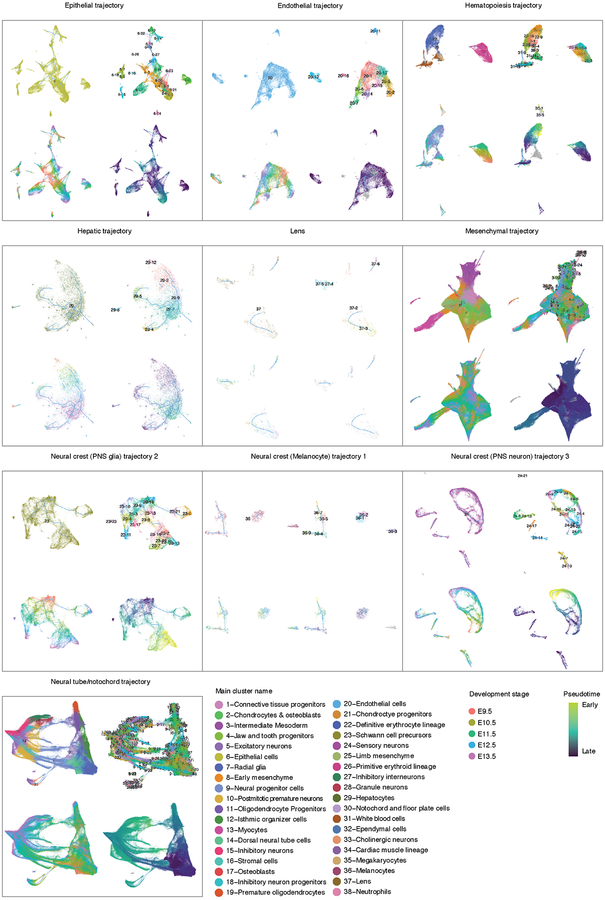
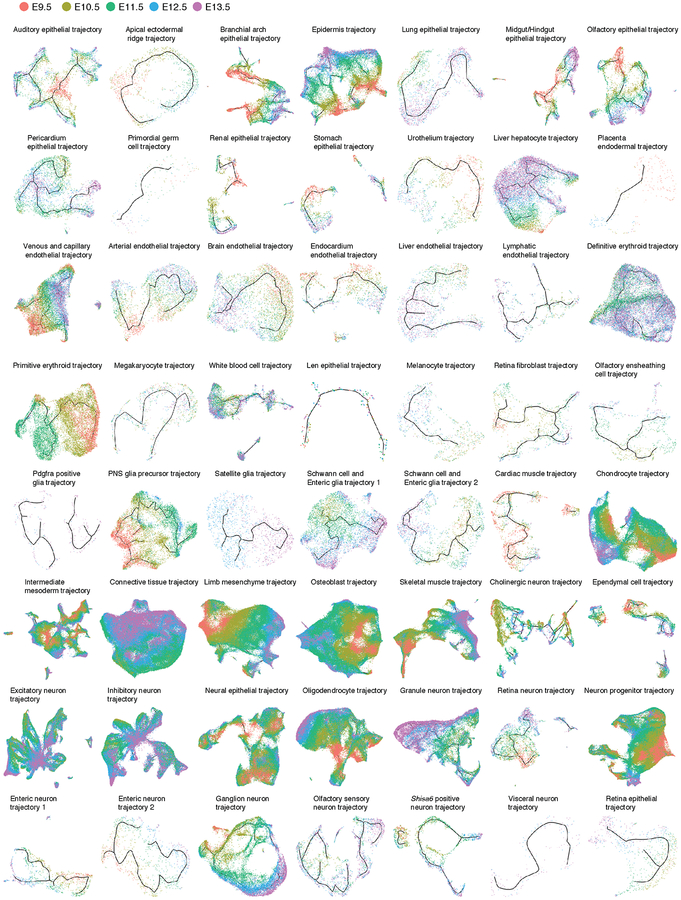
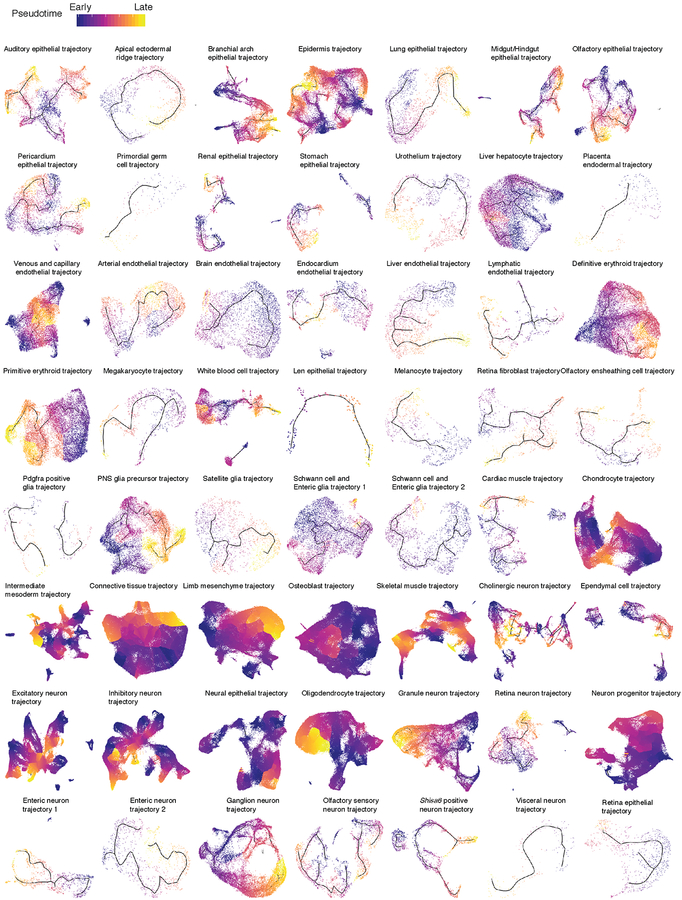
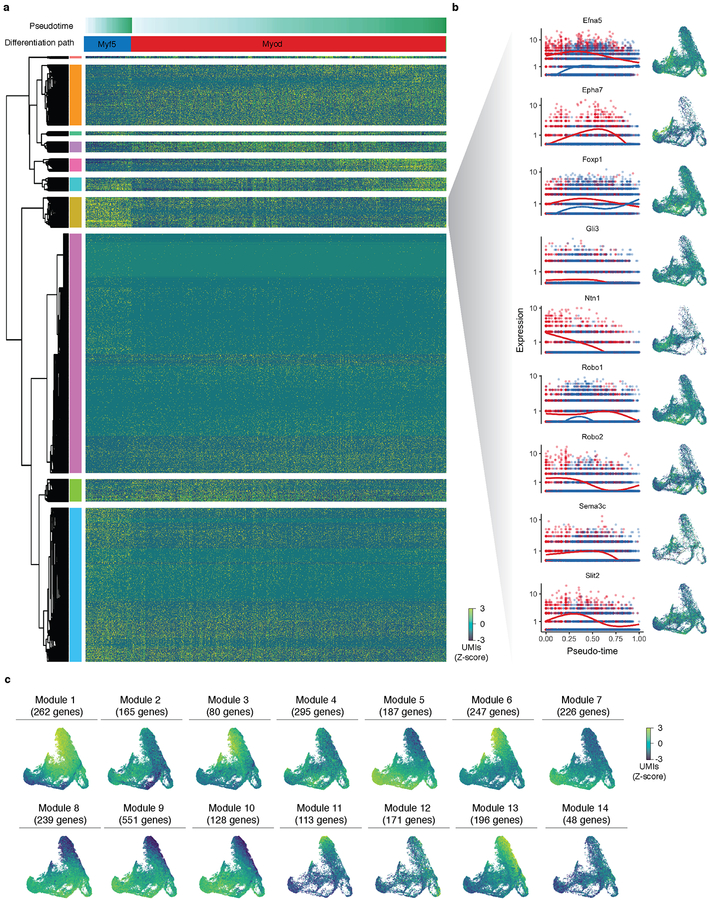
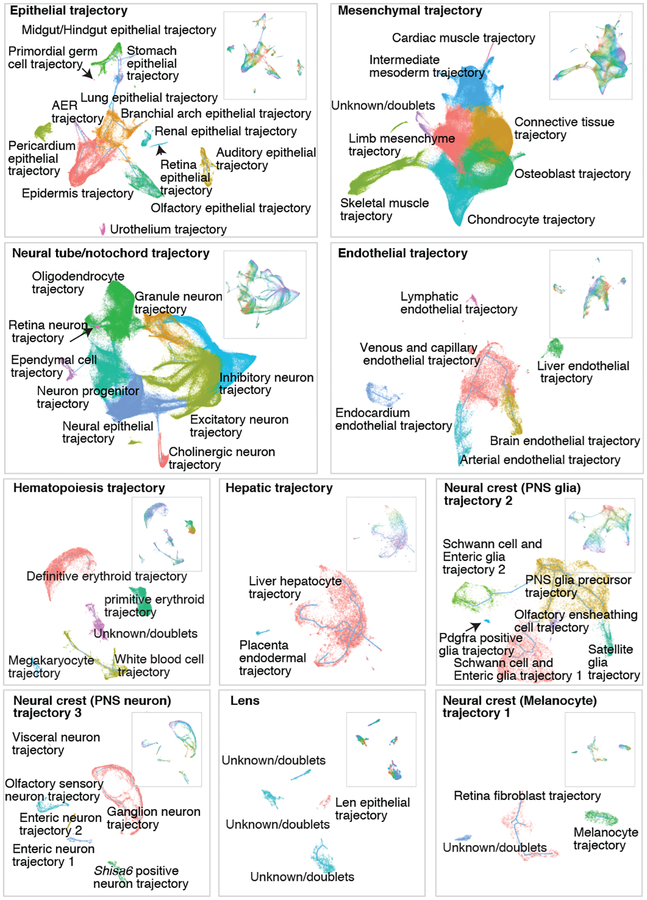
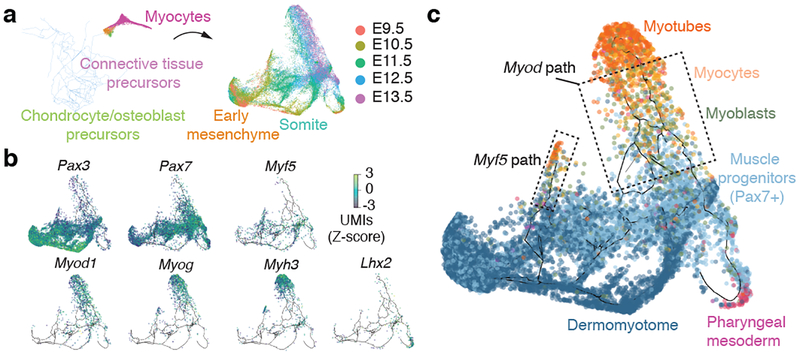
Mammalian organogenesis is a remarkable process. Within a short timeframe, the cells of the three germ layers transform into an embryo that includes most of the major internal and external organs. Here we investigate the transcriptional dynamics of mouse organogenesis at single-cell resolution. Using single-cell combinatorial indexing, we profiled the transcriptomes of around 2 million cells derived from 61 embryos staged between 9.5 and 13.5 days of gestation, in a single experiment. The resulting 'mouse organogenesis cell atlas' (MOCA) provides a global view of developmental processes during this critical window. We use Monocle 3 to identify hundreds of cell types and 56 trajectories, many of which are detected only because of the depth of cellular coverage, and collectively define thousands of corresponding marker genes. We explore the dynamics of gene expression within cell types and trajectories over time, including focused analyses of the apical ectodermal ridge, limb mesenchyme and skeletal muscle.
Figures
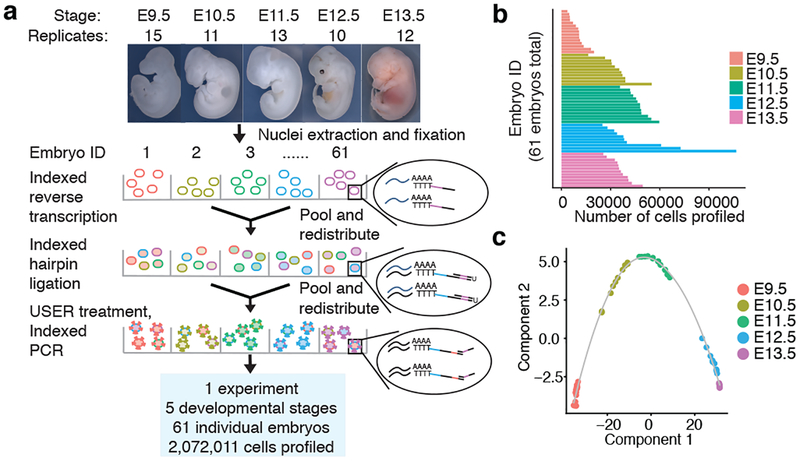
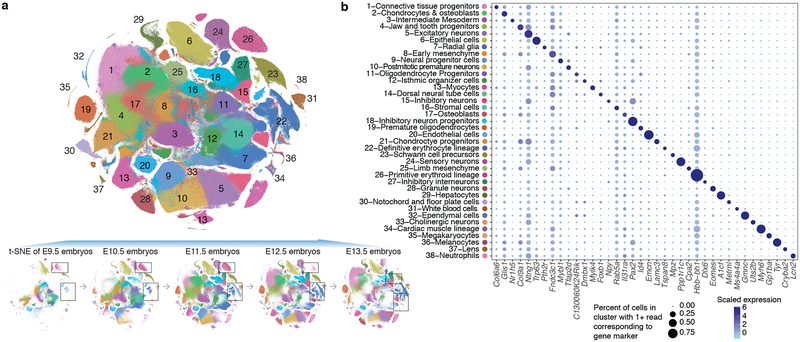
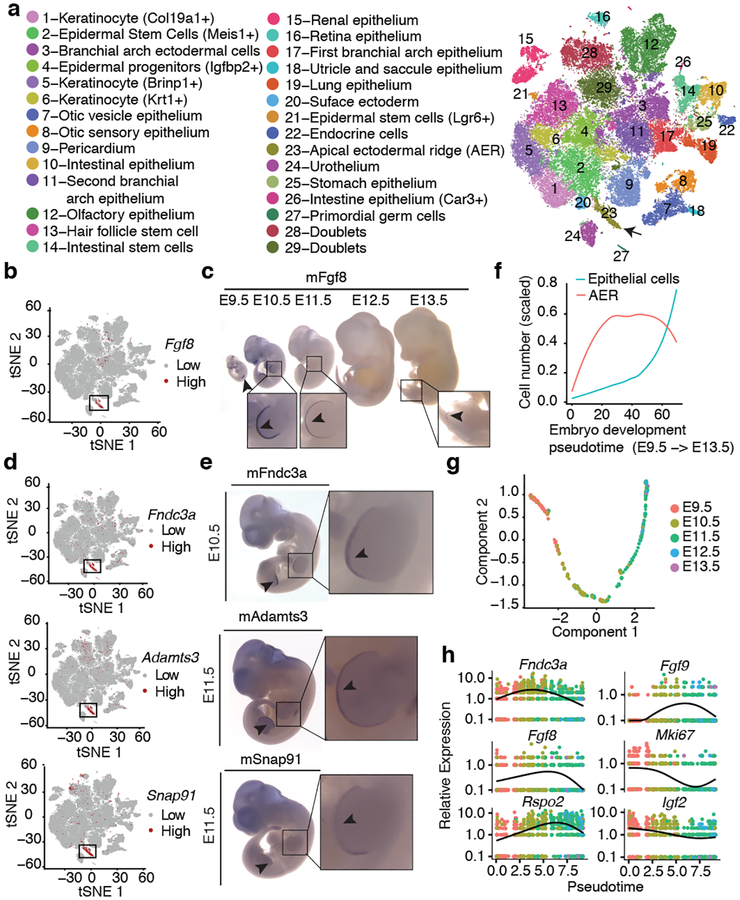
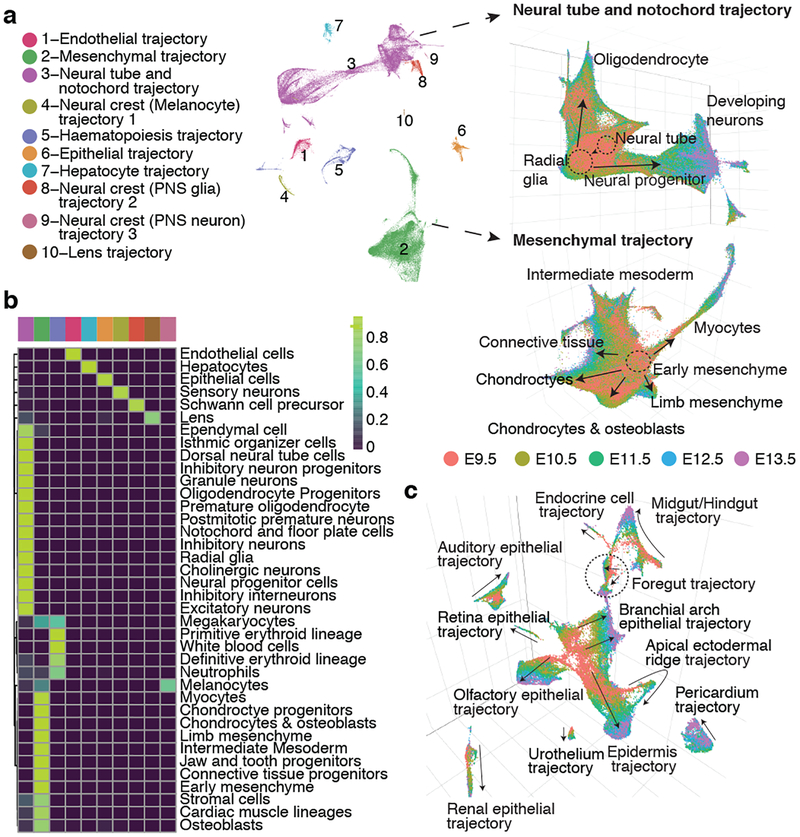
References
Methods References
-
- Li D et al. Formation of proximal and anterior limb skeleton requires early function of Irx3 and Irx5 and is negatively regulated by Shh signaling. Dev. Cell 29, 233–240 (2014). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
