

A novel dominant-negative FGFR1 variant causes Hartsfield syndrome by deregulating RAS/ERK1/2 pathway
- PMID: 30787447
- PMCID: PMC6777633
- DOI: 10.1038/s41431-019-0350-4
A novel dominant-negative FGFR1 variant causes Hartsfield syndrome by deregulating RAS/ERK1/2 pathway
Abstract
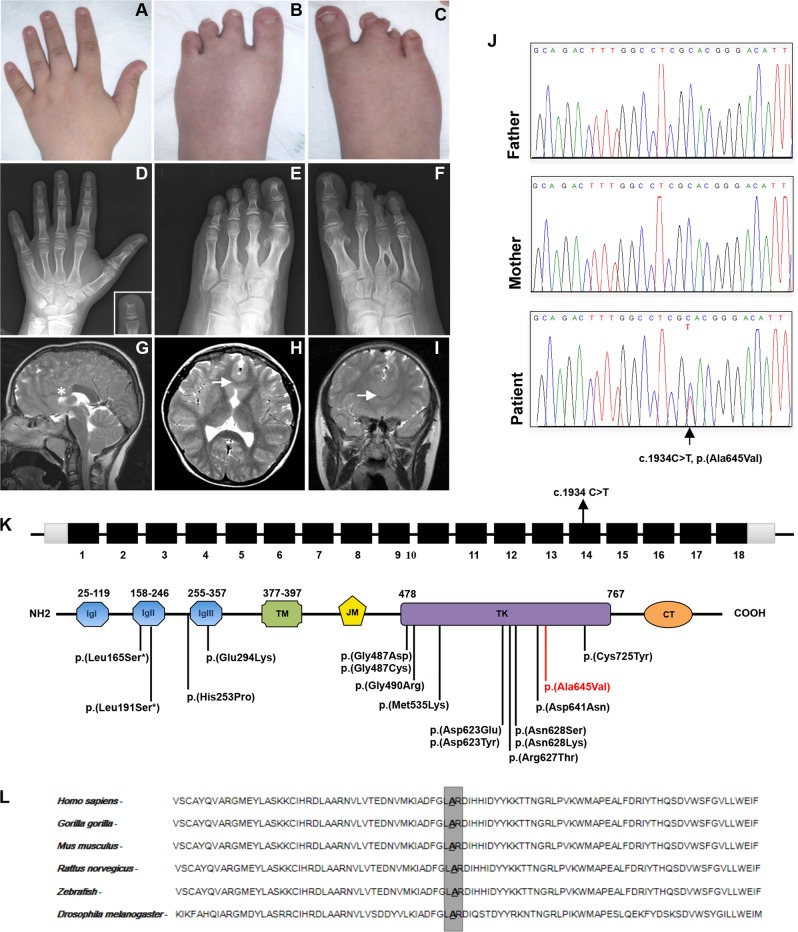
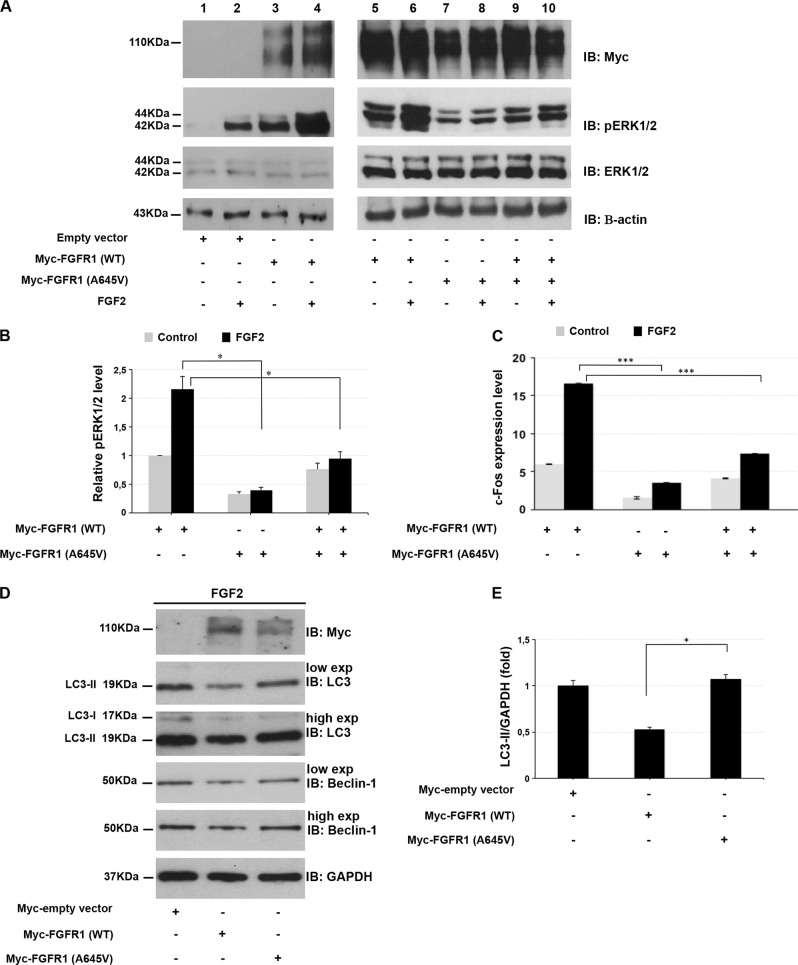
Hartsfield syndrome (HS) is an ultrarare developmental disorder mainly featuring holoprosencephaly and ectrodactyly. It is caused by heterozygous or biallelic variants in FGFR1. Recently, a dominant-negative effect was suggested for FGFR1 variants associated with HS. Here, exome sequencing analysis in a 12-year-old boy with HS disclosed a novel de novo heterozygous variant c.1934C>T in FGFR1 predicted to cause the p.(Ala645Val) amino-acid substitution. In order to evaluate whether the variant, changing a highly conserved residue of the kinase domain, affects FGFR1 function, biochemical studies were employed. We measured the FGFR1 receptor activity in FGF2-treated cell lines exogenously expressing wild-type or Ala645Val FGFR1 by monitoring the activation status of FGF2/FGFR1 downstream pathways. Our analysis highlighted that RAS/ERK1/2 signaling was significantly perturbed in cells expressing mutated FGFR1, in comparison with control cells. We also provided preliminary evidence showing a modulation of the autophagic process in cells expressing mutated FGFR1. This study expands the FGFR1 mutational spectrum associated with HS, provides functional evidence further supporting a dominant-negative effect of this category of FGFR1 variants and offers initial insights on dysregulation of autophagy in HS.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Dhamija R, Babovic-Vuksanovic D. Hartsfield syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2018 (last updated: 3 Mar 2016).
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
