

Neuron-glia interactions in the pathophysiology of epilepsy
- PMID: 30792501
- PMCID: PMC8558781
- DOI: 10.1038/s41583-019-0126-4
Neuron-glia interactions in the pathophysiology of epilepsy
Abstract
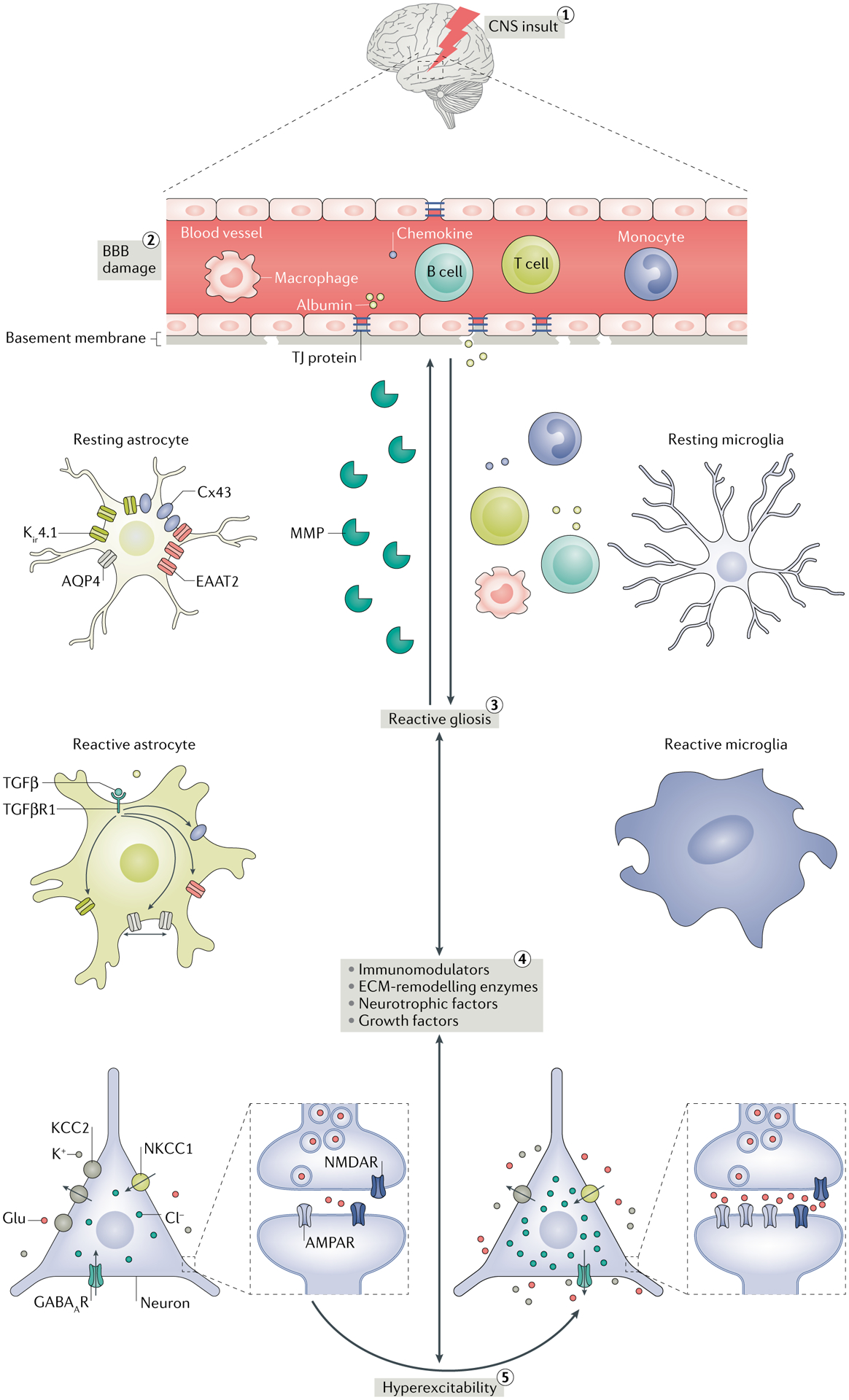
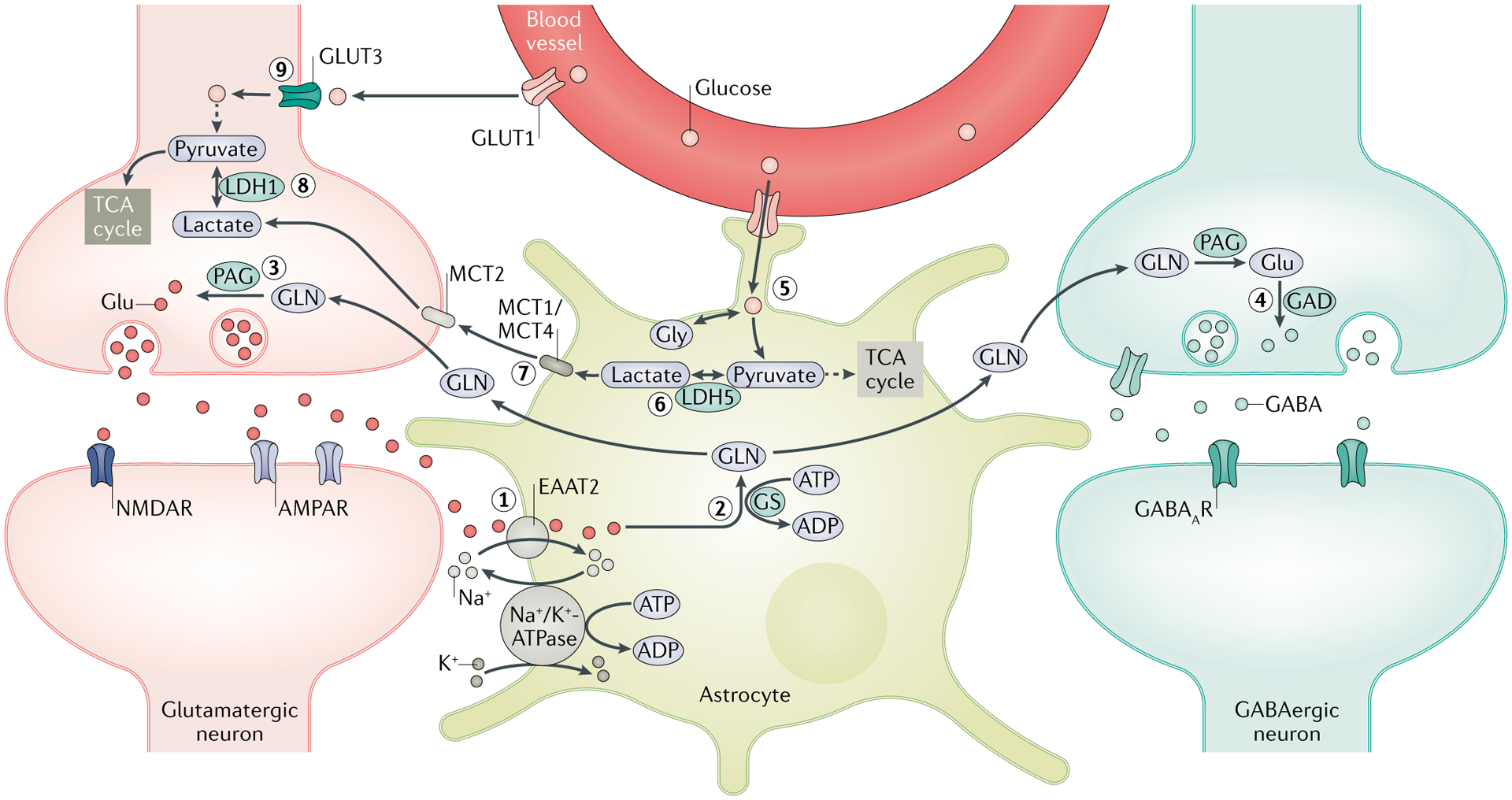
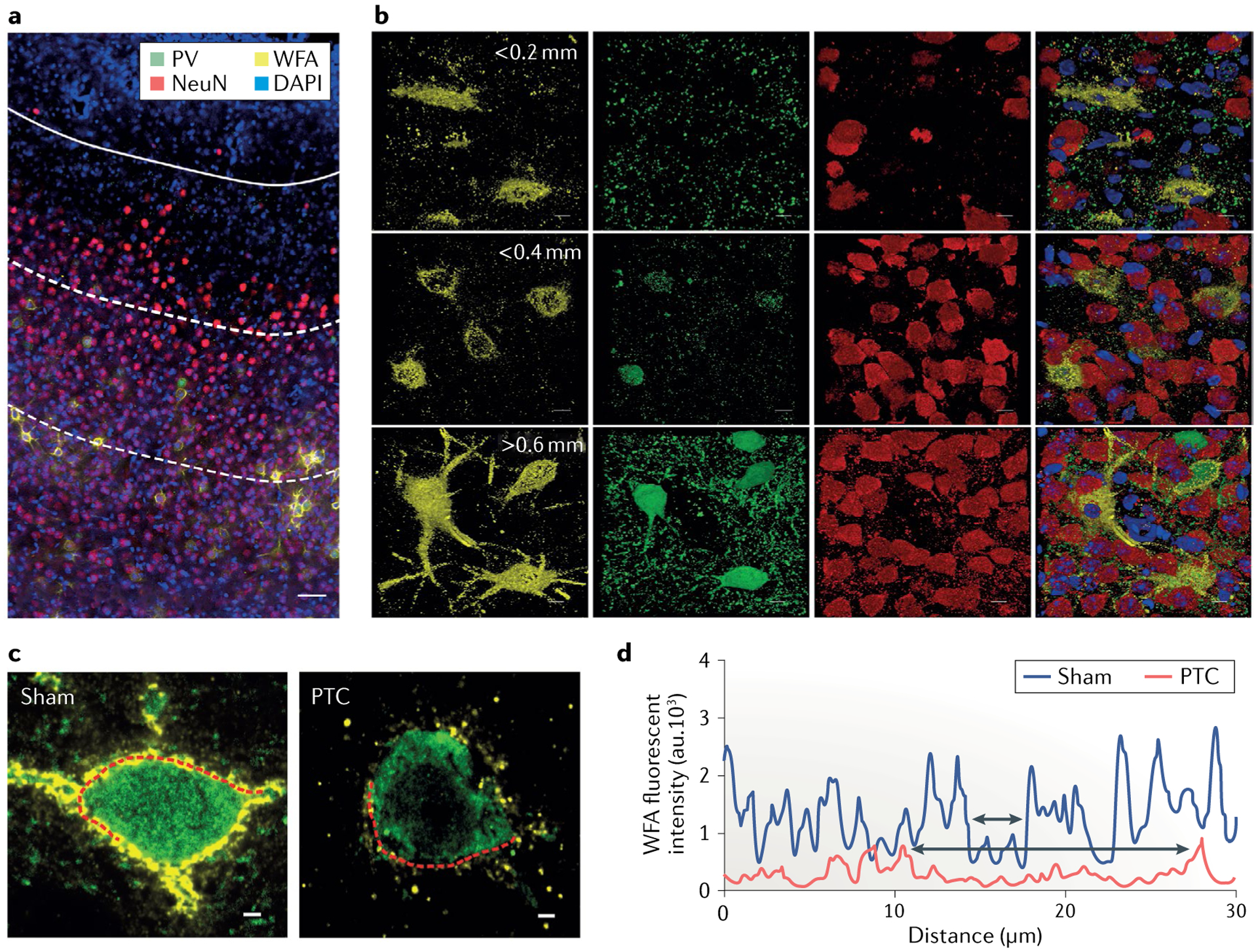
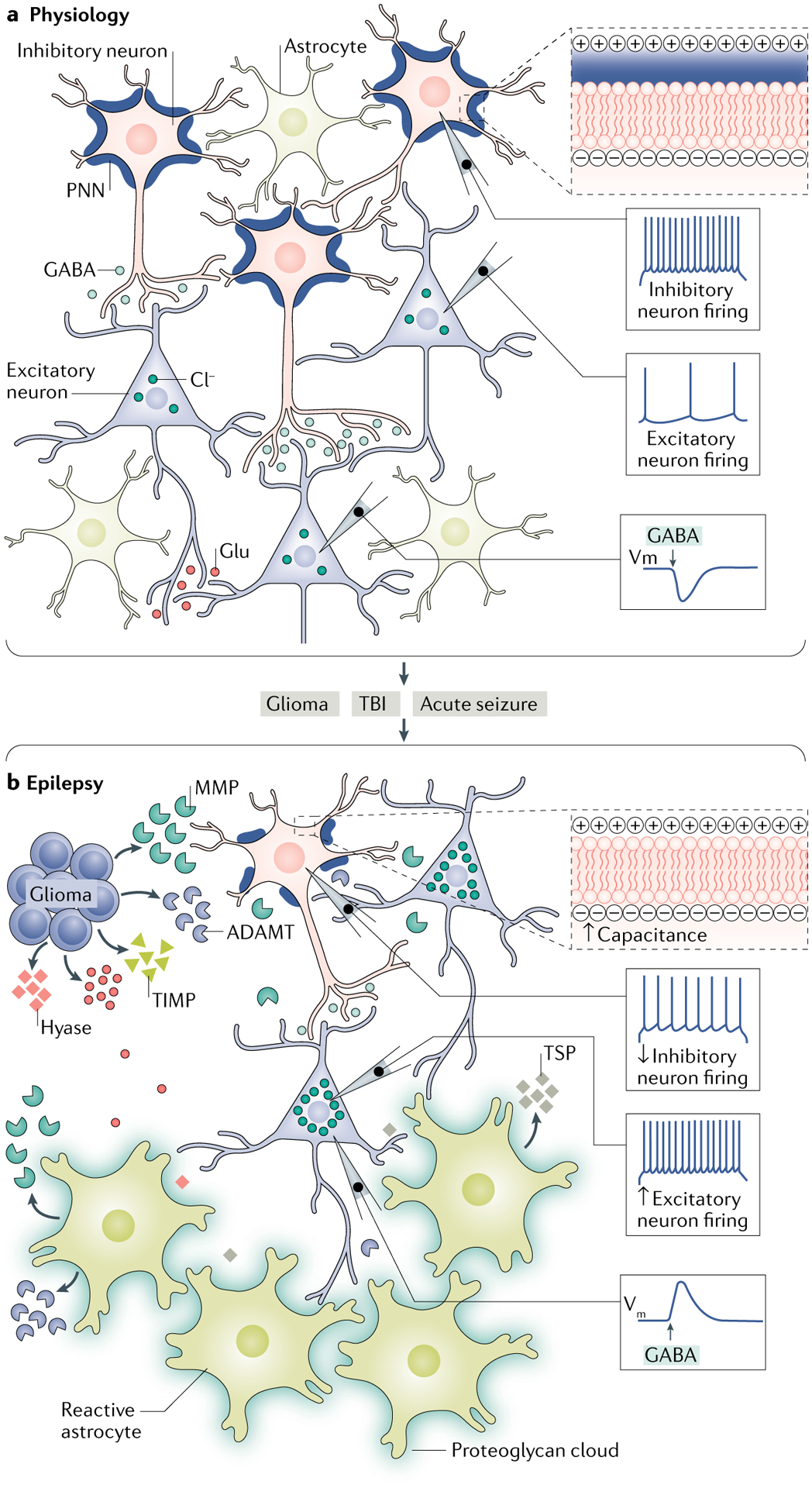
Epilepsy is a neurological disorder afflicting ~65 million people worldwide. It is caused by aberrant synchronized firing of populations of neurons primarily due to imbalance between excitatory and inhibitory neurotransmission. Hence, the historical focus of epilepsy research has been neurocentric. However, the past two decades have enjoyed an explosion of research into the role of glia in supporting and modulating neuronal activity, providing compelling evidence of glial involvement in the pathophysiology of epilepsy. The mechanisms by which glia, particularly astrocytes and microglia, may contribute to epilepsy and consequently could be harnessed therapeutically are discussed in this Review.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- Sontheimer H Diseases of the Nervous System 61–95 (Elsevier, 2015).
-
- Fisher RS et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 55, 475–482 (2014). - PubMed
-
- Loscher W, Hirsch LJ & Schmidt D The enigma of the latent period in the development of symptomatic acquired epilepsy - traditional view versus new concepts. Epilepsy Behav.52, 78–92 (2015). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
