

Mechanisms Underlying Neuroprotection by the NSAID Mefenamic Acid in an Experimental Model of Stroke
- PMID: 30792624
- PMCID: PMC6374636
- DOI: 10.3389/fnins.2019.00064
Mechanisms Underlying Neuroprotection by the NSAID Mefenamic Acid in an Experimental Model of Stroke
Abstract
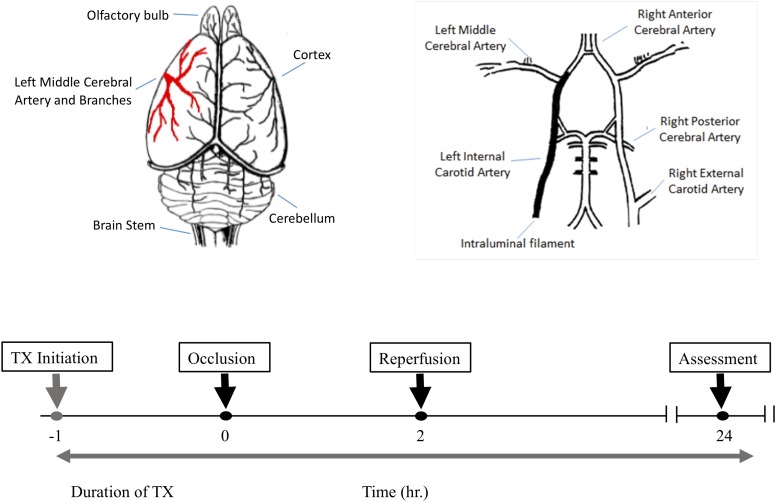
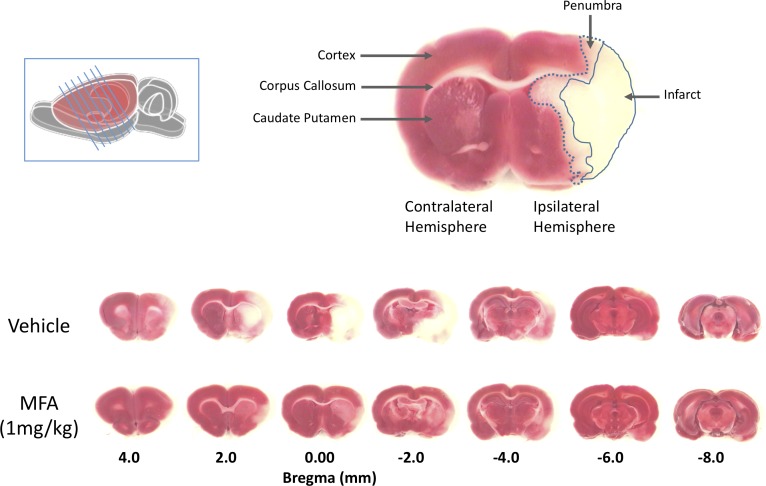
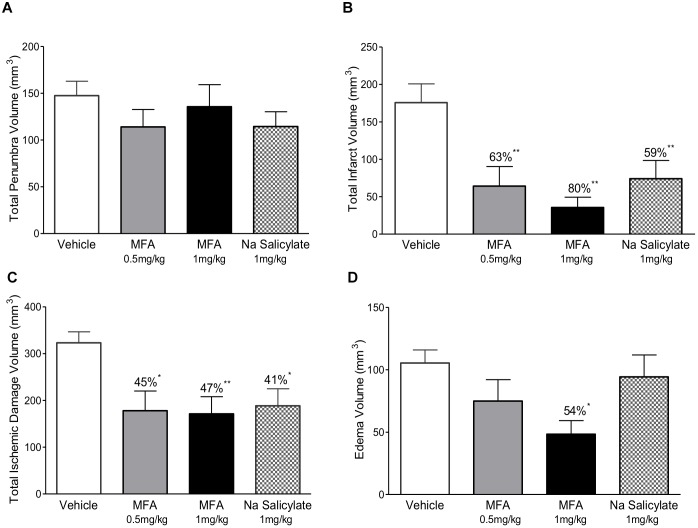
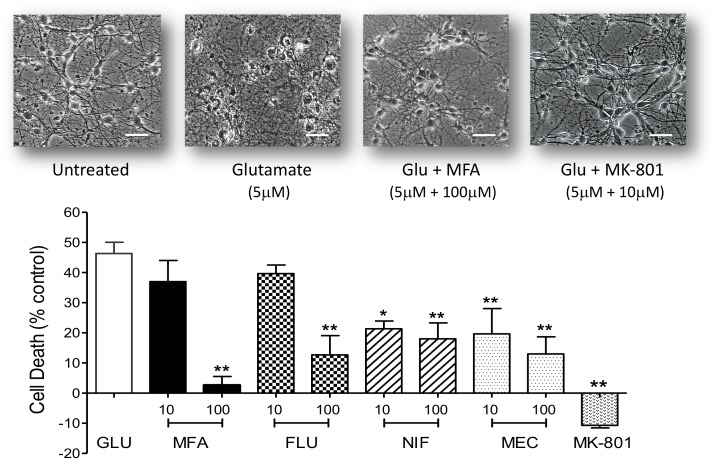
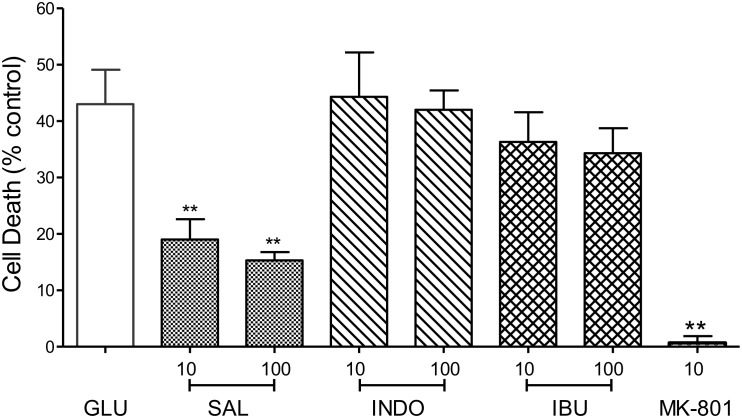
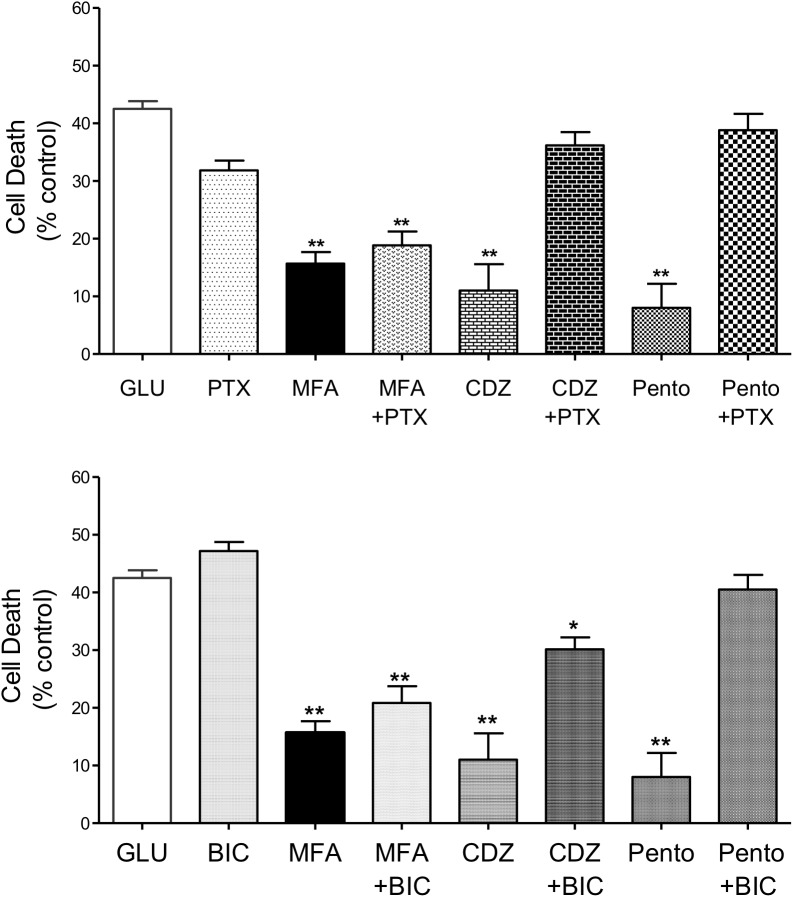
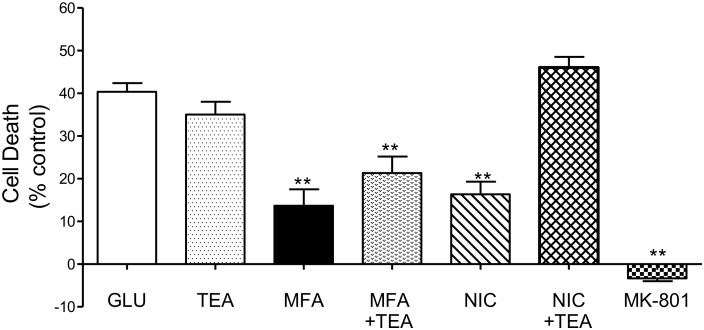
Stroke is a devastating neurological event with limited treatment opportunities. Recent advances in understanding the underlying pathogenesis of cerebral ischemia support the involvement of multiple biochemical pathways in the development of the ischemic damage. Fenamates are classical non-steroidal anti-inflammatory drugs but they are also highly subunit-selective modulators of GABAA receptors, activators of IKS potassium channels and antagonists of non-selective cation channels and the NLRP3 inflammosome. In the present study we investigated the effect of mefenamic acid (MFA) in a rodent model of ischemic stroke and then addressed the underlying pharmacological mechanisms in vitro for its actions in vivo. The efficacy of MFA in reducing ischemic damage was evaluated in adult male Wistar rats subjected to a 2-h middle cerebral artery occlusion. Intracerebroventricular (ICV) infusion of MFA (0.5 or 1 mg/kg) for 24 h, significantly reduced the infarct volume and the total ischemic brain damage. In vitro, the fenamates, MFA, meclofenamic acid, niflumic acid, and flufenamic acid each reduced glutamate-evoked excitotoxicity in cultured embryonic rat hippocampal neurons supporting the idea that this is a drug class action. In contrast the non-fenamate NSAIDs, ibuprofen and indomethacin did not reduce excitotoxicity in vitro indicating that neuroprotection by MFA was not dependent upon anti-inflammatory actions. Co-application of MFA (100 μM) with either of the GABAA antagonists picrotoxin (100 μM) or bicuculline (10 μM) or the potassium channel blocker tetraethylammonium (30 mM) did not prevent neuroprotection with MFA, suggesting that the actions of MFA also do not depend on GABAA receptor modulation or potassium channel activation. These new findings indicate that fenamates may be valuable in the adjunctive treatment of ischemic stroke.
Keywords: MCAO; excitotoxicity; fenamates; ischemic stroke; neuroprotection.
Figures
Similar articles
-
Antiseizure properties of fenamate NSAIDs determined in mature human stem-cell derived neuroglial circuits.Front Pharmacol. 2024 May 16;15:1385523. doi: 10.3389/fphar.2024.1385523. eCollection 2024. Front Pharmacol. 2024. PMID: 38828453 Free PMC article.
-
Evidence for neuroprotection by the fenamate NSAID, mefenamic acid.Neurochem Int. 2009 Dec;55(7):683-8. doi: 10.1016/j.neuint.2009.06.014. Epub 2009 Jun 27. Neurochem Int. 2009. PMID: 19563851
-
Multiple actions of fenamates and other nonsteroidal anti-inflammatory drugs on GABAA receptors.Eur J Pharmacol. 2019 Jun 15;853:247-255. doi: 10.1016/j.ejphar.2019.03.039. Epub 2019 Mar 28. Eur J Pharmacol. 2019. PMID: 30930251
-
Degradation of fenamates.Profiles Drug Subst Excip Relat Methodol. 2025;50:229-275. doi: 10.1016/bs.podrm.2024.09.001. Epub 2024 Nov 2. Profiles Drug Subst Excip Relat Methodol. 2025. PMID: 39855777 Review.
-
[Neuroprotective actions of lithium].Seishin Shinkeigaku Zasshi. 2003;105(1):81-6. Seishin Shinkeigaku Zasshi. 2003. PMID: 12701214 Review. Japanese.
Cited by
-
Potential Drug Candidates to Treat TRPC6 Channel Deficiencies in the Pathophysiology of Alzheimer's Disease and Brain Ischemia.Cells. 2020 Oct 24;9(11):2351. doi: 10.3390/cells9112351. Cells. 2020. PMID: 33114455 Free PMC article. Review.
-
Mitochondrial protective effects caused by the administration of mefenamic acid in sepsis.J Neuroinflammation. 2022 Nov 4;19(1):268. doi: 10.1186/s12974-022-02616-6. J Neuroinflammation. 2022. PMID: 36333747 Free PMC article.
-
Therapeutic Strategies for Immune Transformation in Parkinson's Disease.J Parkinsons Dis. 2022;12(s1):S201-S222. doi: 10.3233/JPD-223278. J Parkinsons Dis. 2022. PMID: 35871362 Free PMC article. Review.
-
Antiseizure properties of fenamate NSAIDs determined in mature human stem-cell derived neuroglial circuits.Front Pharmacol. 2024 May 16;15:1385523. doi: 10.3389/fphar.2024.1385523. eCollection 2024. Front Pharmacol. 2024. PMID: 38828453 Free PMC article.
-
Mechanism of Curcuma longa and Its Neuroactive Components for the Management of Epileptic Seizures: A Systematic Review.Curr Neuropharmacol. 2021;19(9):1496-1518. doi: 10.2174/1570159X19666210517120413. Curr Neuropharmacol. 2021. PMID: 33998991 Free PMC article.
References
LinkOut - more resources
Full Text Sources