

Using long-read sequencing to detect imprinted DNA methylation
- PMID: 30793194
- PMCID: PMC6486641
- DOI: 10.1093/nar/gkz107
Using long-read sequencing to detect imprinted DNA methylation
Abstract
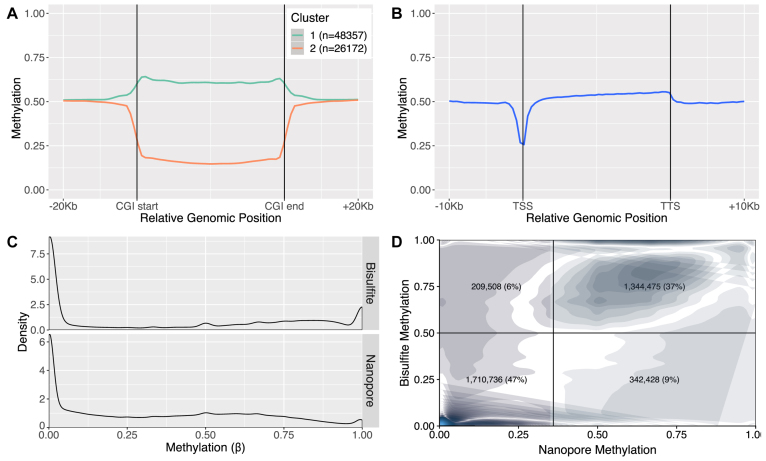
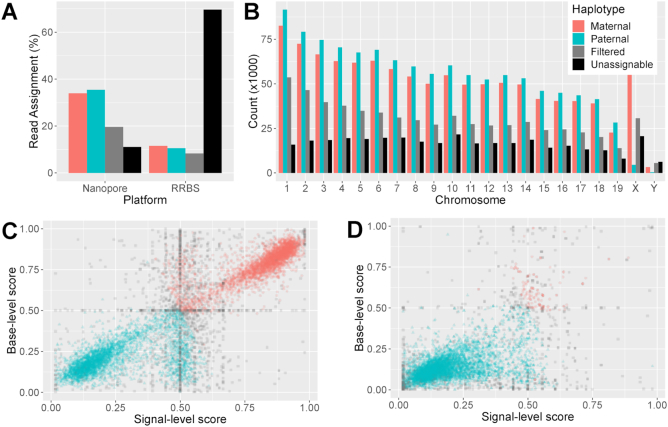
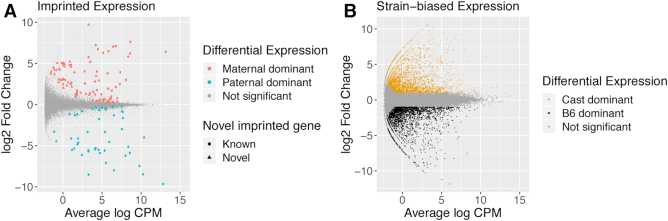
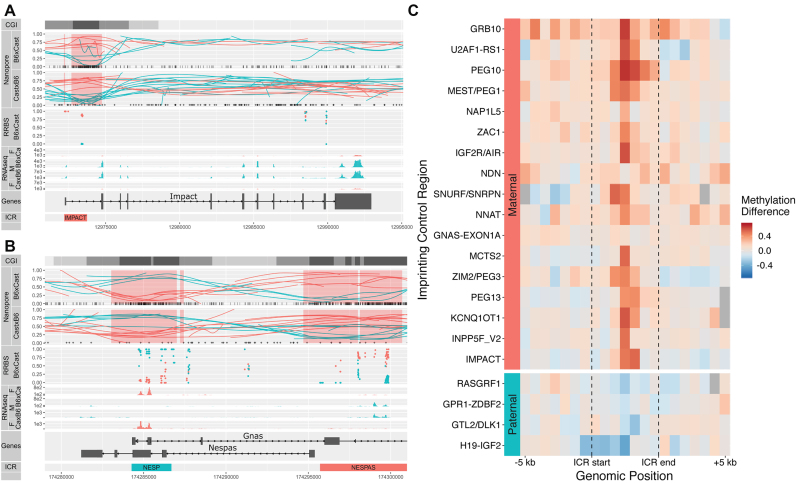
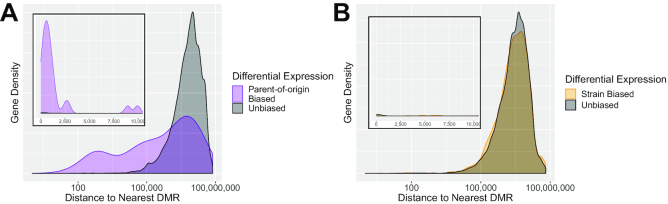
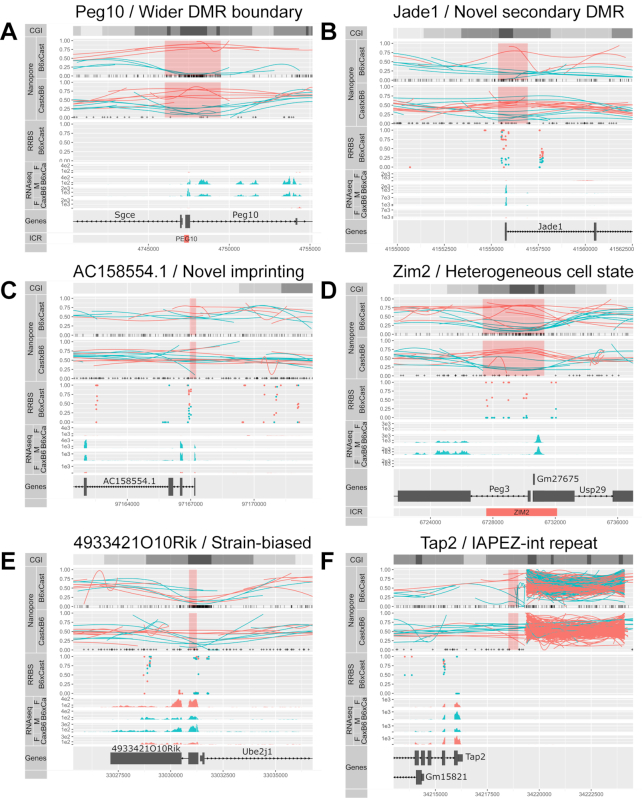
Systematic variation in the methylation of cytosines at CpG sites plays a critical role in early development of humans and other mammals. Of particular interest are regions of differential methylation between parental alleles, as these often dictate monoallelic gene expression, resulting in parent of origin specific control of the embryonic transcriptome and subsequent development, in a phenomenon known as genomic imprinting. Using long-read nanopore sequencing we show that, with an average genomic coverage of ∼10, it is possible to determine both the level of methylation of CpG sites and the haplotype from which each read arises. The long-read property is exploited to characterize, using novel methods, both methylation and haplotype for reads that have reduced basecalling precision compared to Sanger sequencing. We validate the analysis both through comparison of nanopore-derived methylation patterns with those from Reduced Representation Bisulfite Sequencing data and through comparison with previously reported data. Our analysis successfully identifies known imprinting control regions (ICRs) as well as some novel differentially methylated regions which, due to their proximity to hitherto unknown monoallelically expressed genes, may represent new ICRs.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Ferguson-Smith A.C., Sasaki H., Cattanach B.M., Surani M.A.. Parental-origin-specific epigenetic modification of the mouse H19 gene. Nature. 1993; 362:751–755. - PubMed
-
- Stöger R., Kubicka P., Liu C.G., Kafri T., Razin A., Cedar H., Barlow D.P.. Maternal-specific methylation of the imprinted mouse Igf2r locus identifies the expressed locus as carrying the imprinting signal. Cell. 1993; 73:61–71. - PubMed
-
- Bartolomei M.S., Webber A.L., Brunkow M.E., Tilghman S.M.. Epigenetic mechanisms underlying the imprinting of the mouse H19 gene. Genes Dev. 1993; 7:1663–1673. - PubMed
-
- Li E., Beard C., Jaenisch R.. Role for DNA methylation in genomic imprinting. Nature. 1993; 366:362–365. - PubMed
-
- Ferguson-Smith A.C. Genomic imprinting: the emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011; 12:565–575. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
