

CDKL5 deficiency predisposes neurons to cell death through the deregulation of SMAD3 signaling
- PMID: 30793413
- PMCID: PMC8028508
- DOI: 10.1111/bpa.12716
CDKL5 deficiency predisposes neurons to cell death through the deregulation of SMAD3 signaling
Erratum in
-
Corrigendum.Brain Pathol. 2020 May;30(3):721. doi: 10.1111/bpa.12827. Epub 2020 Feb 14. Brain Pathol. 2020. PMID: 32385961 Free PMC article. No abstract available.
Abstract
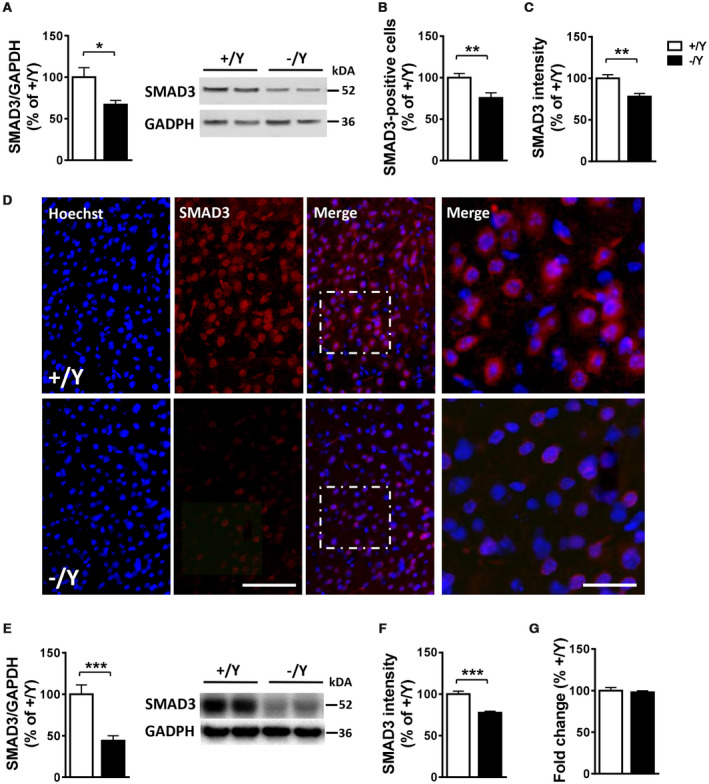
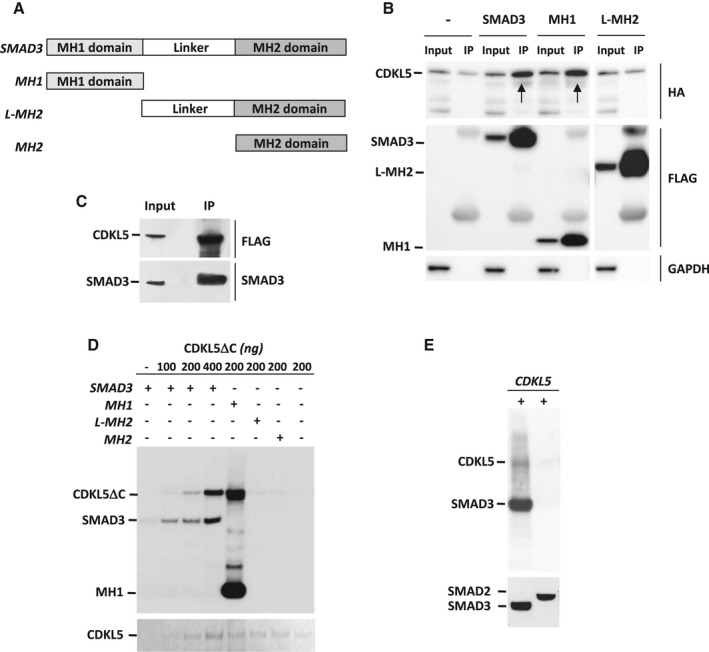
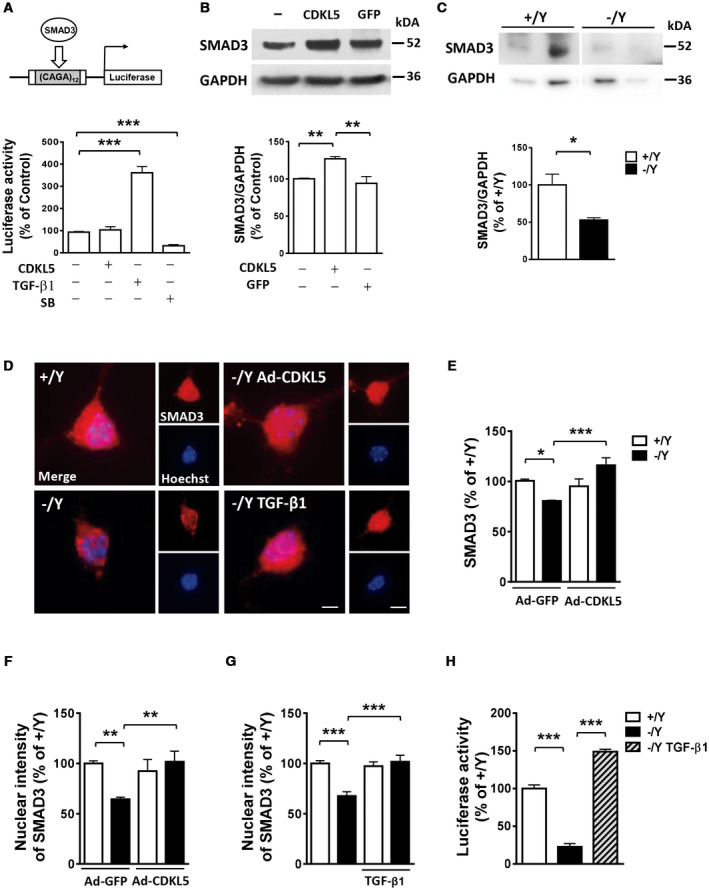
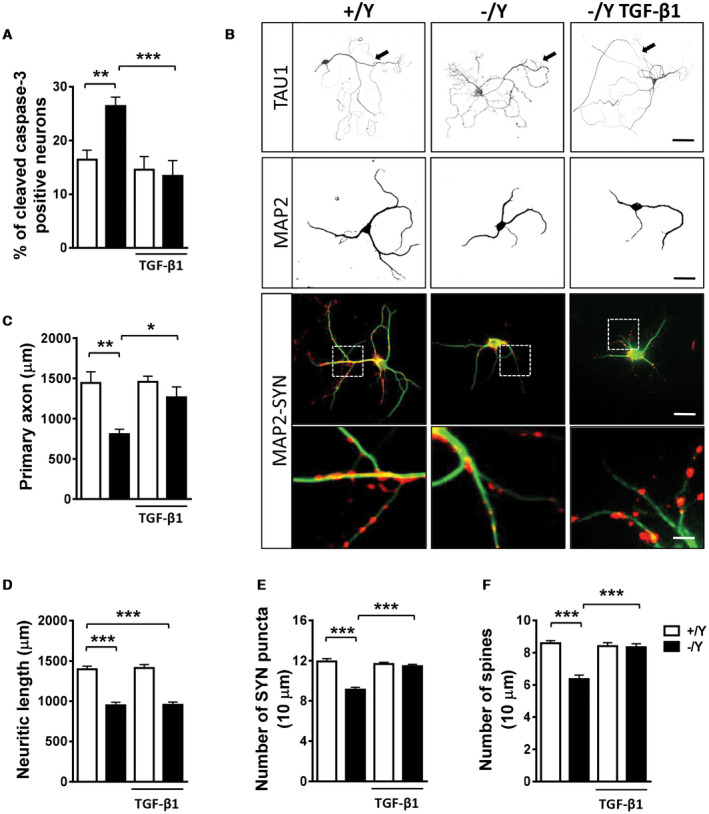
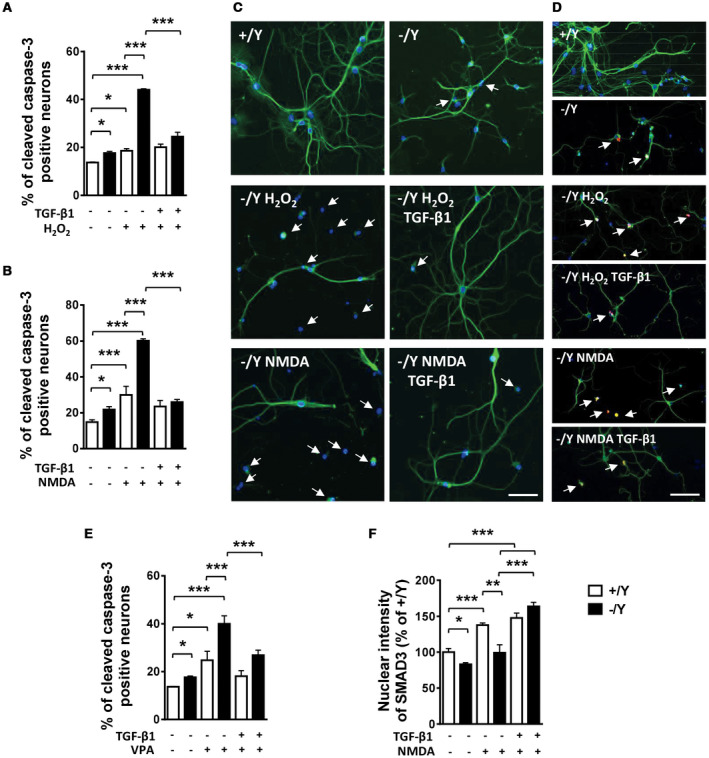
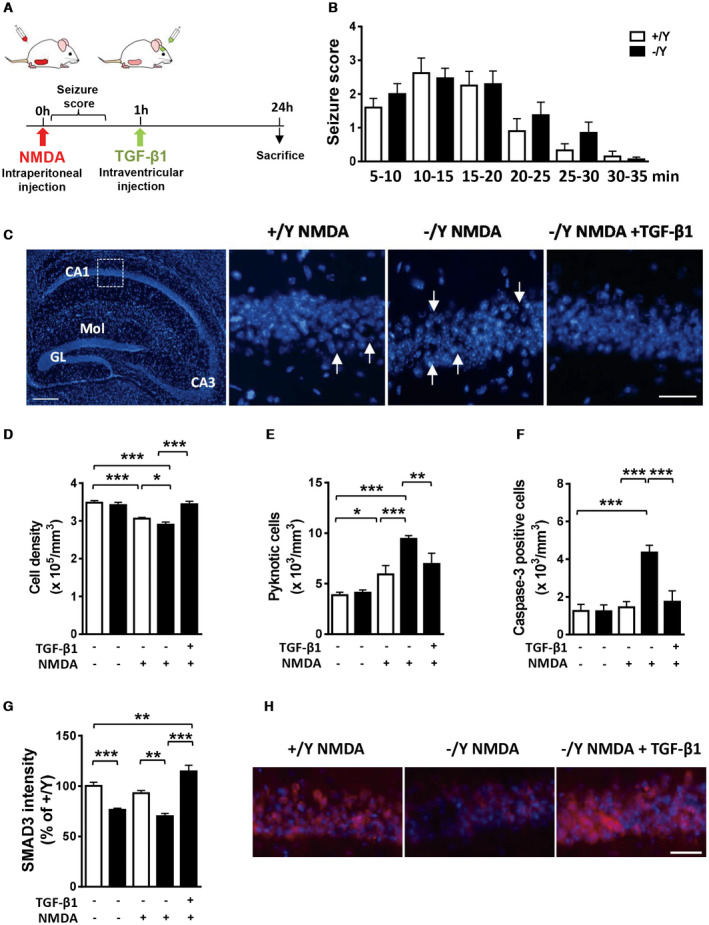
CDKL5 deficiency disorder (CDD) is a rare encephalopathy characterized by early onset epilepsy and severe intellectual disability. CDD is caused by mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5) gene, a member of a highly conserved family of serine-threonine kinases. Only a few physiological substrates of CDKL5 are currently known, which hampers the discovery of therapeutic strategies for CDD. Here, we show that SMAD3, a primary mediator of TGF-β action, is a direct phosphorylation target of CDKL5 and that CDKL5-dependent phosphorylation promotes SMAD3 protein stability. Importantly, we found that restoration of the SMAD3 signaling through TGF-β1 treatment normalized defective neuronal survival and maturation in Cdkl5 knockout (KO) neurons. Moreover, we demonstrate that Cdkl5 KO neurons are more vulnerable to neurotoxic/excitotoxic stimuli. In vivo treatment with TGF-β1 prevents increased NMDA-induced cell death in hippocampal neurons from Cdkl5 KO mice, suggesting an involvement of the SMAD3 signaling deregulation in the neuronal susceptibility to excitotoxic injury of Cdkl5 KO mice. Our finding reveals a new function for CDKL5 in maintaining neuronal survival that could have important implications for susceptibility to neurodegeneration in patients with CDD.
Keywords: CDKL5; SMAD3; TGF-β signaling; hippocampal neurons; neuronal maturation; neuronal survival.
© 2019 International Society of Neuropathology.
Conflict of interest statement
All authors reported that they had no biomedical financial interests or potential conflicts of interest.
Figures
Similar articles
-
Microtubules: A Key to Understand and Correct Neuronal Defects in CDKL5 Deficiency Disorder?Int J Mol Sci. 2019 Aug 21;20(17):4075. doi: 10.3390/ijms20174075. Int J Mol Sci. 2019. PMID: 31438497 Free PMC article. Review.
-
The green tea polyphenol epigallocatechin-3-gallate (EGCG) restores CDKL5-dependent synaptic defects in vitro and in vivo.Neurobiol Dis. 2020 May;138:104791. doi: 10.1016/j.nbd.2020.104791. Epub 2020 Feb 4. Neurobiol Dis. 2020. PMID: 32032735 Free PMC article.
-
Inhibition of microglia overactivation restores neuronal survival in a mouse model of CDKL5 deficiency disorder.J Neuroinflammation. 2021 Jul 8;18(1):155. doi: 10.1186/s12974-021-02204-0. J Neuroinflammation. 2021. PMID: 34238328 Free PMC article.
-
Cyclin-Dependent Kinase-Like 5 (CDKL5): Possible Cellular Signalling Targets and Involvement in CDKL5 Deficiency Disorder.Neural Plast. 2020 Jun 5;2020:6970190. doi: 10.1155/2020/6970190. eCollection 2020. Neural Plast. 2020. PMID: 32587608 Free PMC article. Review.
-
Increased DNA Damage and Apoptosis in CDKL5-Deficient Neurons.Mol Neurobiol. 2020 May;57(5):2244-2262. doi: 10.1007/s12035-020-01884-8. Epub 2020 Jan 30. Mol Neurobiol. 2020. PMID: 32002787
Cited by
-
Transcriptomic Profiling of Zebrafish Mutant for cdkl5 Reveals Dysregulated Gene Expression Associated with Neuronal, Muscle, Visual and Skeletal Development.Int J Mol Sci. 2025 Jun 24;26(13):6069. doi: 10.3390/ijms26136069. Int J Mol Sci. 2025. PMID: 40649845 Free PMC article.
-
CDKL5 deficiency disorder: molecular insights and mechanisms of pathogenicity to fast-track therapeutic development.Biochem Soc Trans. 2022 Aug 31;50(4):1207-1224. doi: 10.1042/BST20220791. Biochem Soc Trans. 2022. PMID: 35997111 Free PMC article. Review.
-
Microtubules: A Key to Understand and Correct Neuronal Defects in CDKL5 Deficiency Disorder?Int J Mol Sci. 2019 Aug 21;20(17):4075. doi: 10.3390/ijms20174075. Int J Mol Sci. 2019. PMID: 31438497 Free PMC article. Review.
-
The green tea polyphenol epigallocatechin-3-gallate (EGCG) restores CDKL5-dependent synaptic defects in vitro and in vivo.Neurobiol Dis. 2020 May;138:104791. doi: 10.1016/j.nbd.2020.104791. Epub 2020 Feb 4. Neurobiol Dis. 2020. PMID: 32032735 Free PMC article.
-
Insights into the structure and function of the hippocampus: implications for the pathophysiology and treatment of autism spectrum disorder.Front Psychiatry. 2024 Apr 23;15:1364858. doi: 10.3389/fpsyt.2024.1364858. eCollection 2024. Front Psychiatry. 2024. PMID: 38716113 Free PMC article. Review.
References
-
- Barbiero I, Peroni D, Tramarin M, Chandola C, Rusconi L, Landsberger N, Kilstrup‐Nielsen C (2017) The neurosteroid pregnenolone reverts microtubule derangement induced by the loss of a functional CDKL5‐IQGAP1 complex. Hum Mol Gen 26:3520–3530. - PubMed
-
- Bertani I, Rusconi L, Bolognese F, Forlani G, Conca B, De Monte L et al (2006) Functional consequences of mutations in CDKL5, an X‐linked gene involved in infantile spasms and mental retardation. J Biol Chem 281:32048–32056. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Research Materials
