

Angiotensin II-induced hypertension and cardiac hypertrophy are differentially mediated by TLR3- and TLR4-dependent pathways
- PMID: 30793936
- PMCID: PMC6580398
- DOI: 10.1152/ajpheart.00697.2018
Angiotensin II-induced hypertension and cardiac hypertrophy are differentially mediated by TLR3- and TLR4-dependent pathways
Abstract
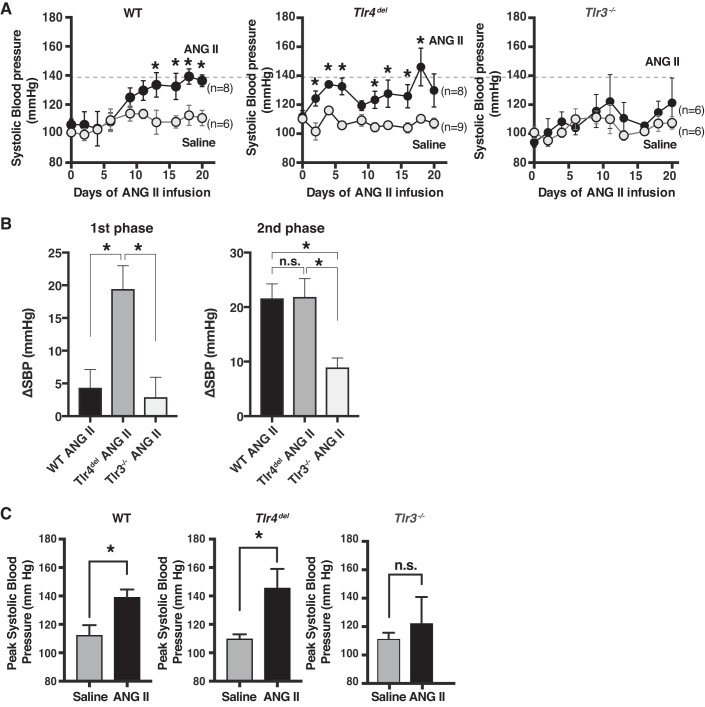
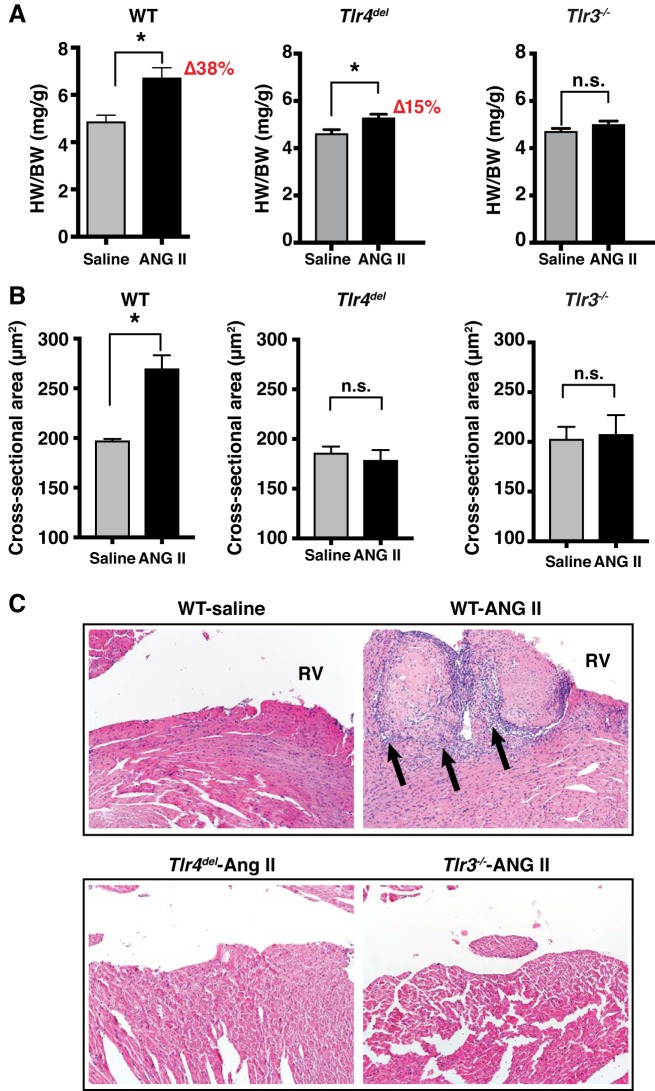
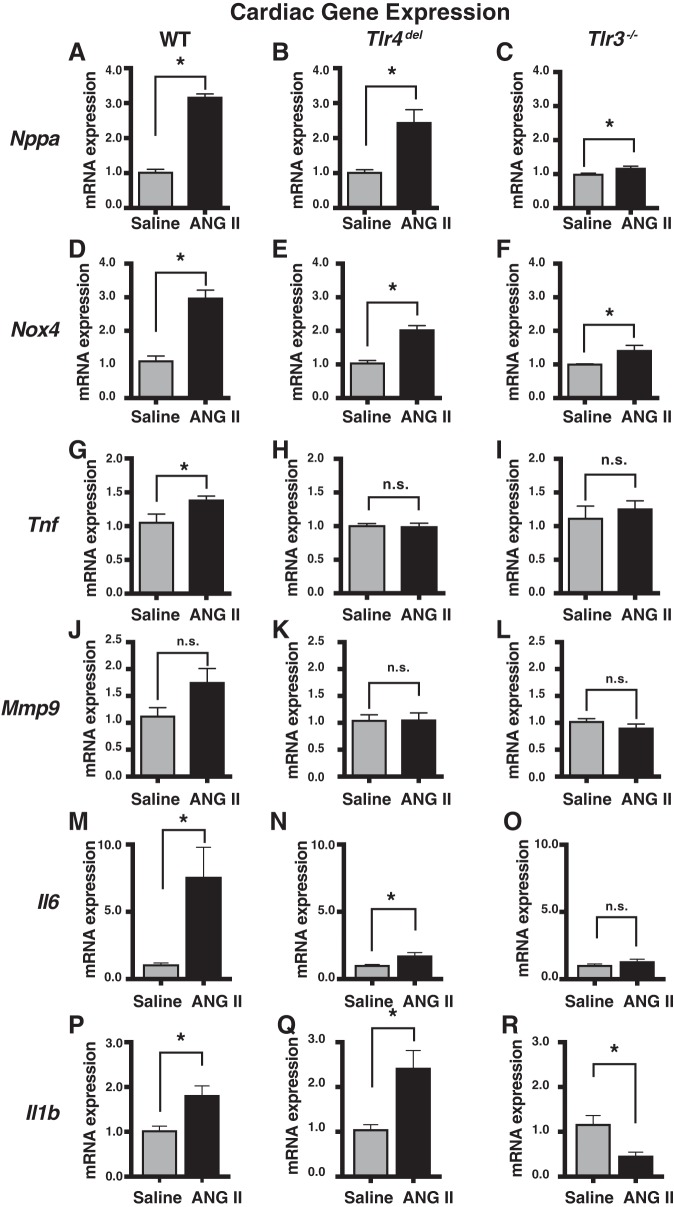
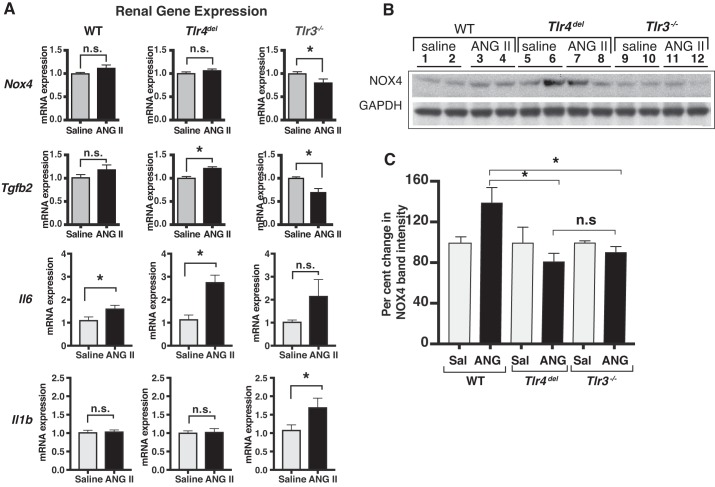
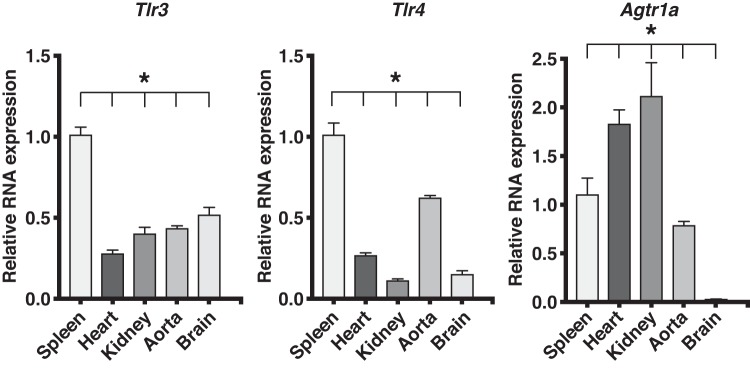
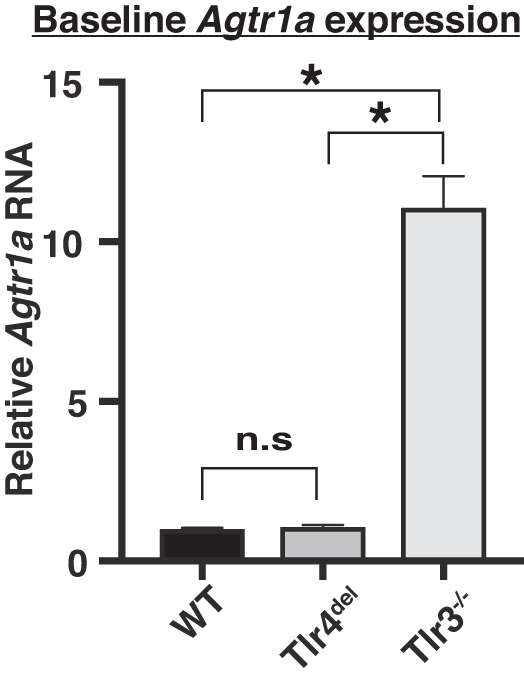
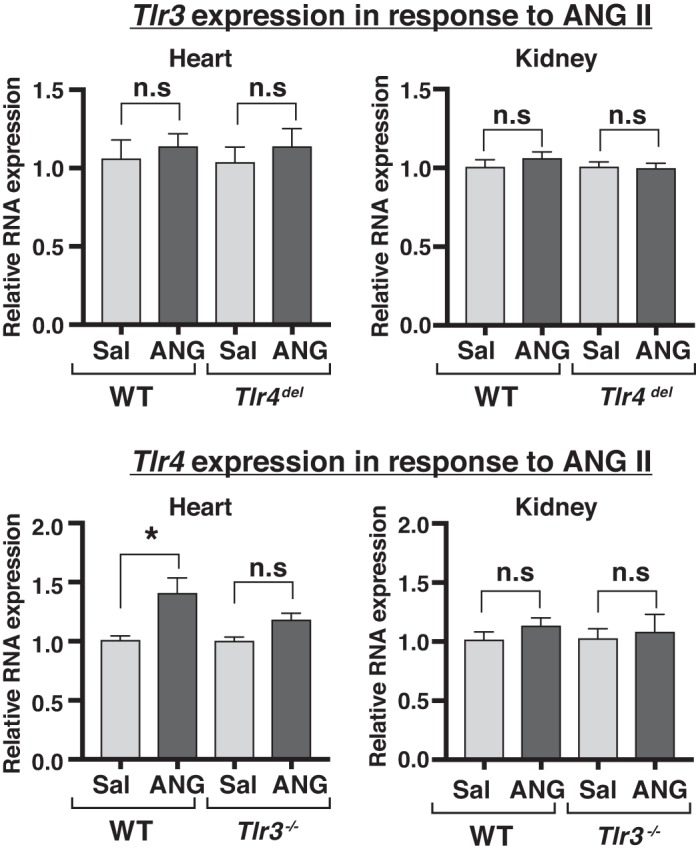
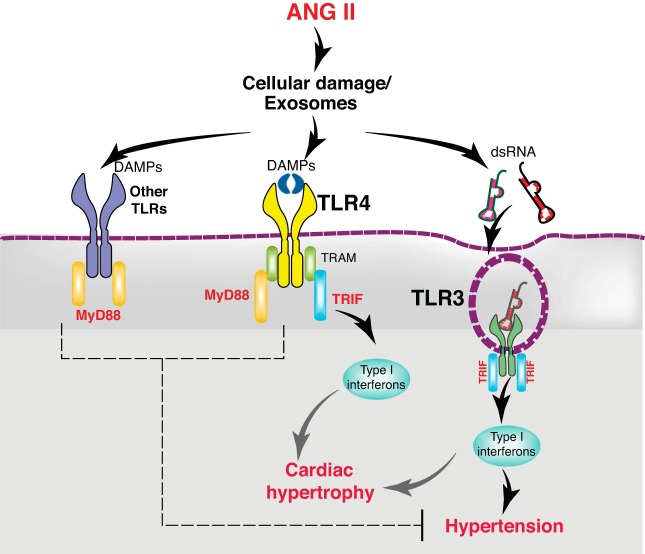
Toll-like receptors (TLR) are key components of the innate immune system that elicit inflammatory responses through the adaptor proteins myeloid differentiation protein 88 (MyD88) and Toll-interleukin receptor domain-containing adaptor protein-inducing interferon-β (TRIF). Previously, we demonstrated that TRIF mediates the signaling of angiotensin II (ANG II)- induced hypertension and cardiac hypertrophy. Since TRIF is activated selectively by TLR3 and TLR4, our goals in this study were to determine the roles of TLR3 and TLR4 in mediating ANG II-induced hypertension and cardiac hypertrophy, and associated changes in proinflammatory gene expression in heart and kidney. In wild-type (WT) mice, ANG II infusion (1,000 ng·kg-1·min-1 for 3 wk) increased systolic blood pressure and caused cardiac hypertrophy. In ANG II-infused TLR4-deficient mice (Tlr4del), hypertrophy was significantly attenuated despite a preserved or enhanced hypertensive response. In contrast, in TLR3-deficient mice (Tlr3-/-), both ANG II-induced hypertension and hypertrophy were abrogated. In WT mice, ANG II increased the expression of several proinflammatory genes in hearts and kidneys that were attenuated in both TLR4- and TLR3-deficient mice compared with WT. We conclude that ANG II activates both TLR4-TRIF and TLR3-TRIF pathways in a nonredundant manner whereby hypertension is dependent on activation of the TLR3-TRIF pathway and cardiac hypertrophy is dependent on both TLR3-TRIF and TLR4-TRIF pathways. NEW & NOTEWORTHY Angiotensin II (ANG II)-induced hypertension is dependent on the endosomal Toll-like receptor 3 (TLR3)-Toll-interleukin receptor domain-containing adaptor protein-inducing interferon-β (TRIF) pathway of the innate immune system but not on cell membrane localized TLR4. However, ANG II-induced cardiac hypertrophy is regulated by both TLR4-TRIF and TLR3-TRIF pathways. Thus, ANG II-induced rise in systolic blood pressure is independent of TLR4-TRIF effect on cardiac hypertrophy. The TLR3-TRIF pathway may be a potential target of therapeutic intervention.
Keywords: MyD88; TLR3; TLR4; TRIF; Toll-like receptors; angiotensin II; cardiac hypertrophy; hypertension; innate immune system.
Figures
References
-
- Bouabout G, Ayme-Dietrich E, Jacob H, Champy MF, Birling MC, Pavlovic G, Madeira L, Fertak LE, Petit-Demoulière B, Sorg T, Herault Y, Mudgett J, Monassier L. Nox4 genetic inhibition in experimental hypertension and metabolic syndrome. Arch Cardiovasc Dis 111: 41–52, 2018. doi: 10.1016/j.acvd.2017.03.011. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous