

Hepcidin-ferroportin axis in health and disease
- PMID: 30798811
- PMCID: PMC7730607
- DOI: 10.1016/bs.vh.2019.01.002
Hepcidin-ferroportin axis in health and disease
Abstract
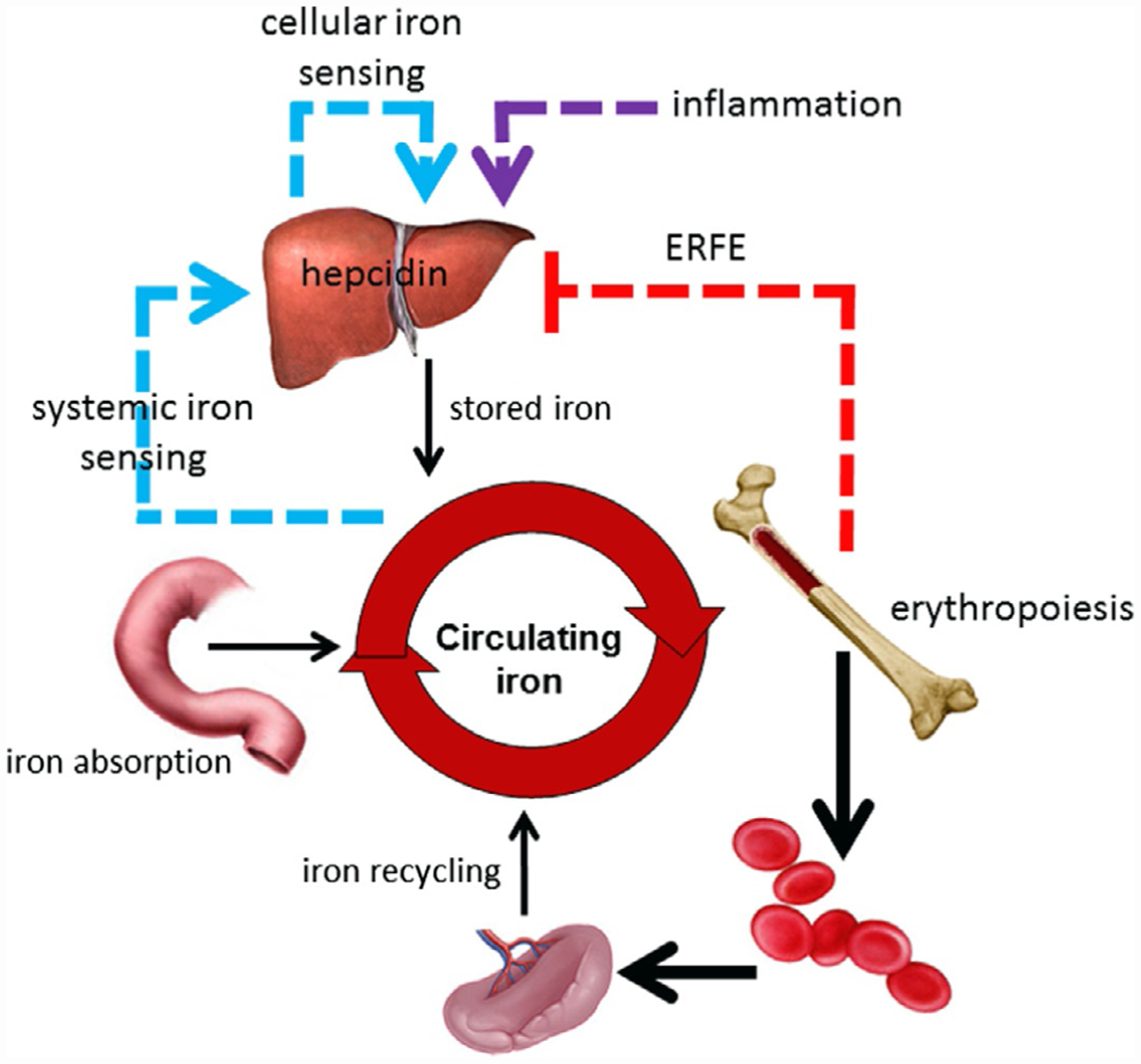
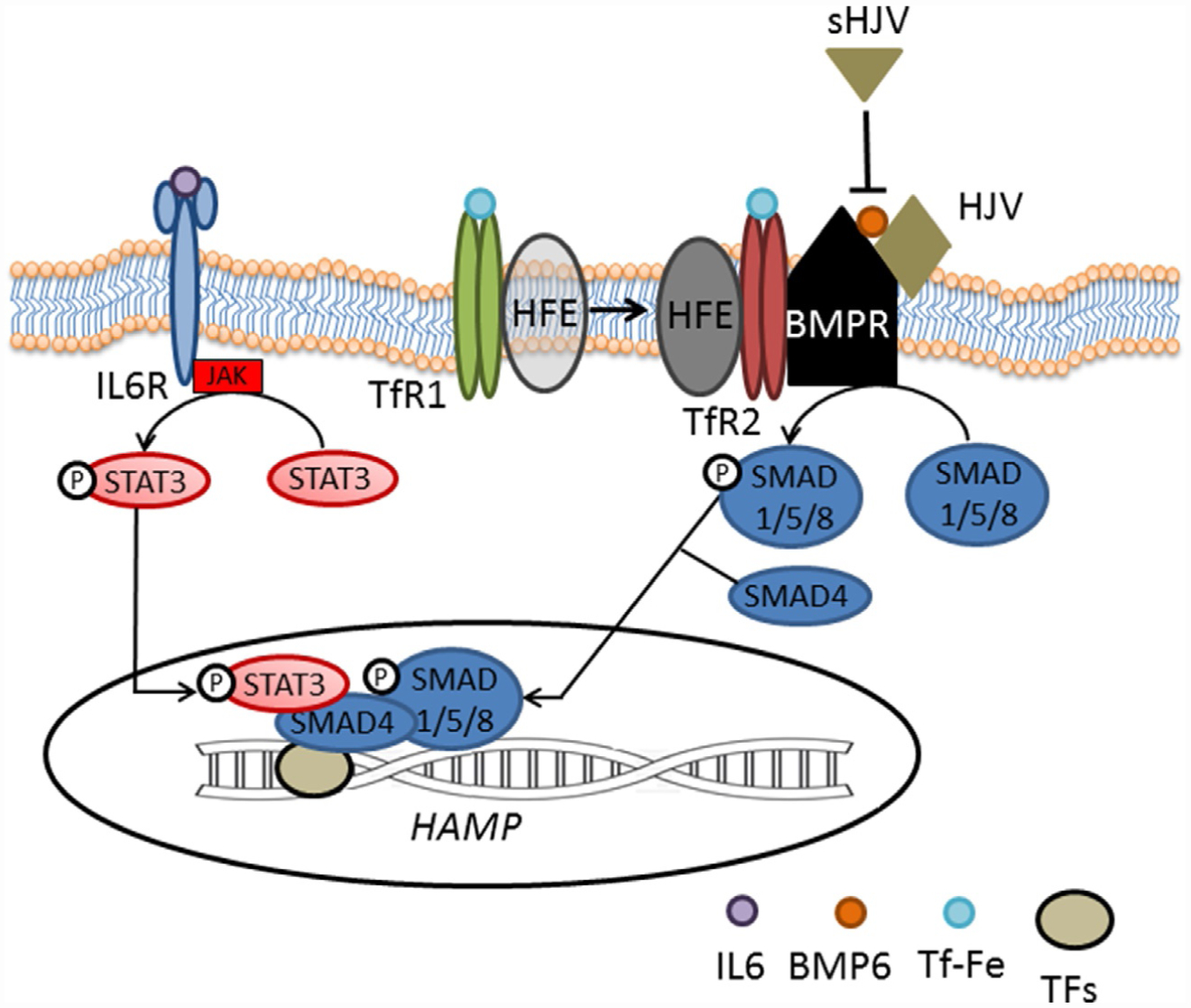
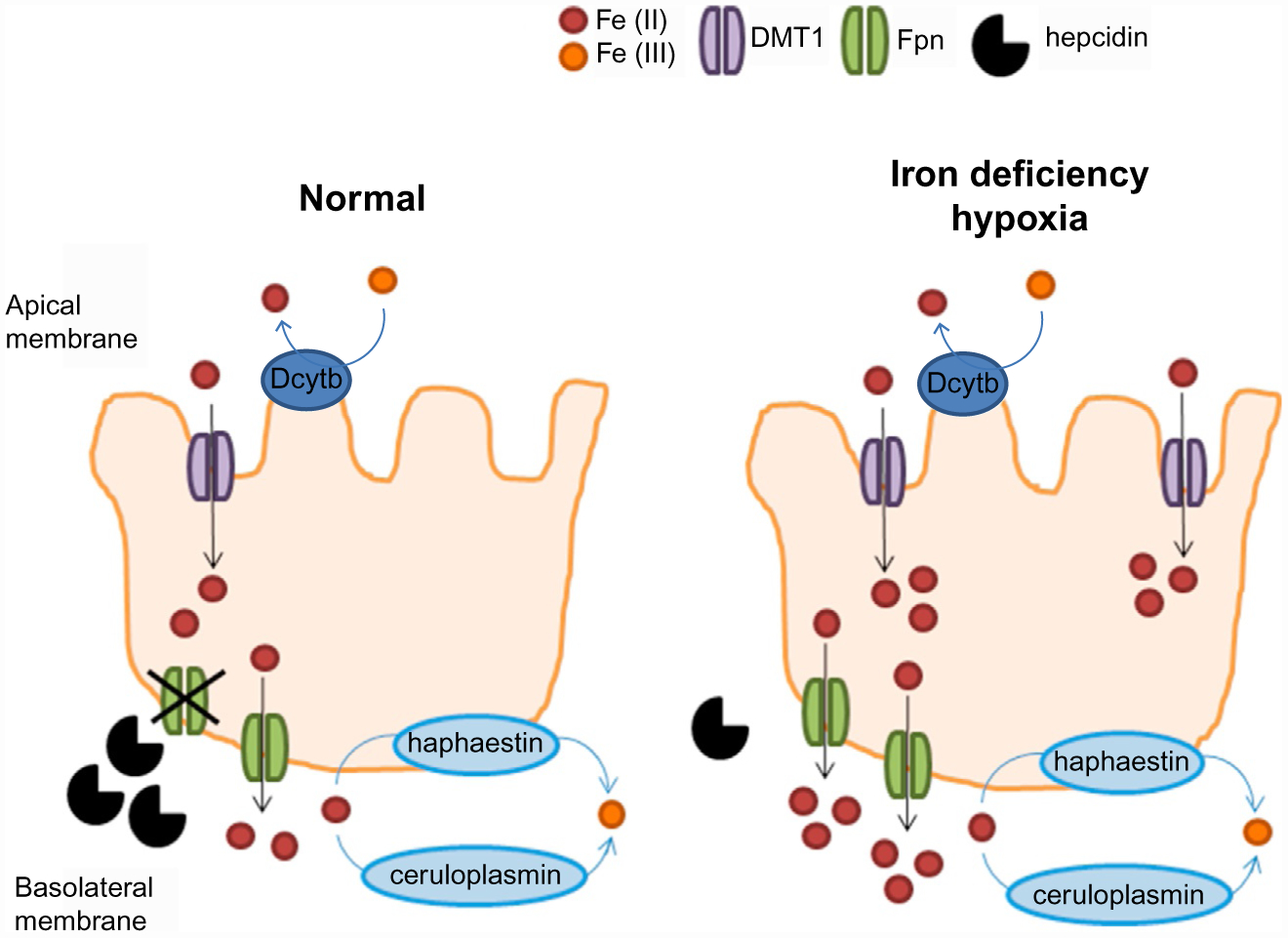
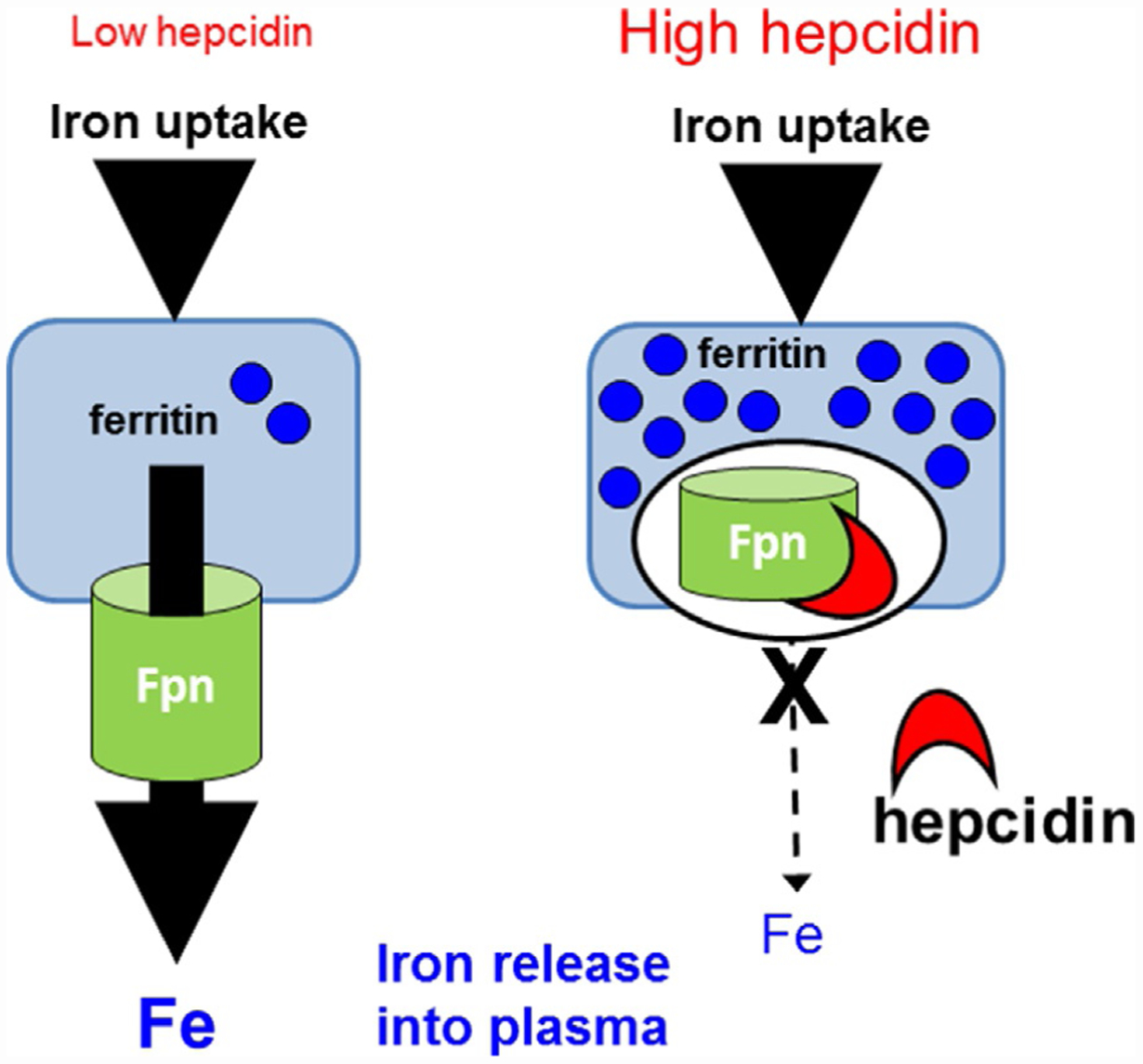
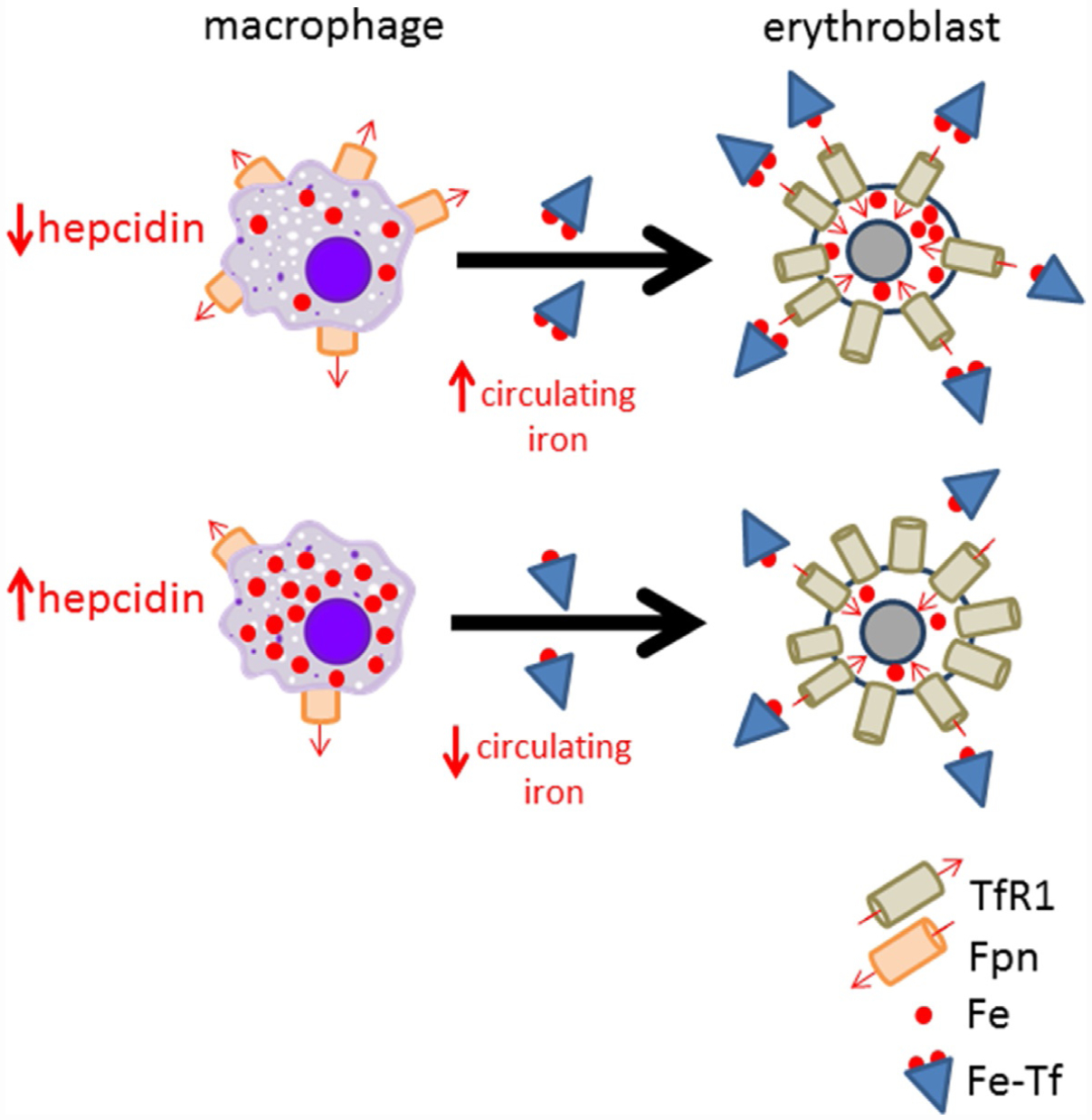
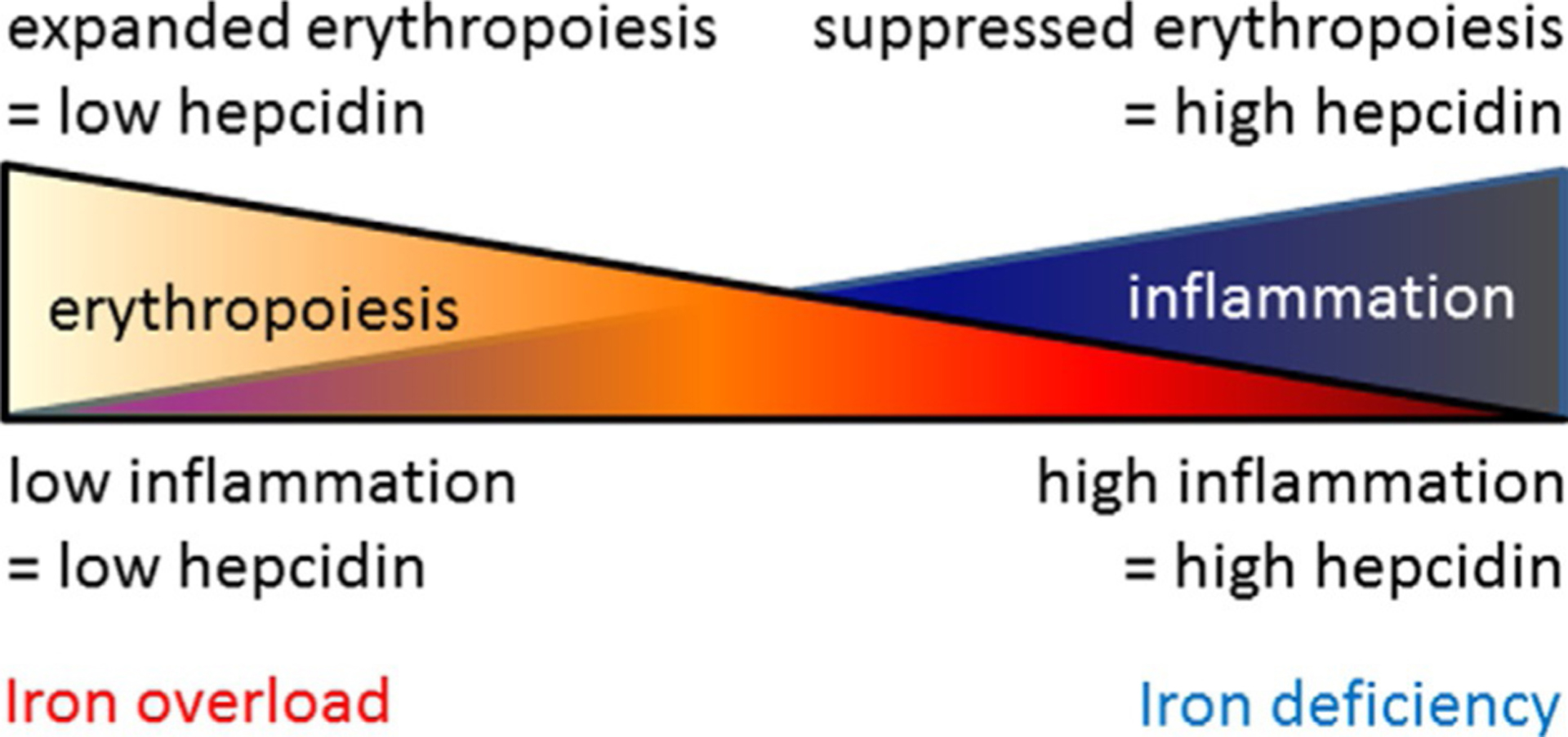
Hepcidin is central to regulation of iron metabolism. Its effect on a cellular level involves binding ferroportin, the main iron export protein, resulting in its internalization and degradation and leading to iron sequestration within ferroportin-expressing cells. Aberrantly increased hepcidin leads to systemic iron deficiency and/or iron restricted erythropoiesis. Furthermore, insufficiently elevated hepcidin occurs in multiple diseases associated with iron overload. Abnormal iron metabolism as a consequence of hepcidin dysregulation is an underlying factor resulting in pathophysiology of multiple diseases and several agents aimed at manipulating this pathway have been designed, with some already in clinical trials. In this chapter, we present an overview of and rationale for exploring the development of hepcidin agonists and antagonists in various clinical scenarios.
Keywords: Anemia of inflammation; Erythroferrone; Hepcidin antagonists; Hepcidin mimetics; Hepcidin:ferroportin axis; Hereditary hemochromatosis; Iron metabolism; Polycythemia vera; β-thalassemia.
© 2019 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Hepcidin and its multiple partners: Complex regulation of iron metabolism in health and disease.Vitam Horm. 2023;123:249-284. doi: 10.1016/bs.vh.2023.03.001. Epub 2023 Mar 30. Vitam Horm. 2023. PMID: 37717987
-
Regulators of hepcidin expression.Vitam Horm. 2019;110:101-129. doi: 10.1016/bs.vh.2019.01.005. Epub 2019 Feb 2. Vitam Horm. 2019. PMID: 30798807 Review.
-
Modulation of hepcidin to treat iron deregulation: potential clinical applications.Expert Rev Hematol. 2016;9(2):169-86. doi: 10.1586/17474086.2016.1124757. Epub 2015 Dec 15. Expert Rev Hematol. 2016. PMID: 26669208 Free PMC article. Review.
-
The dynamics of hepcidin-ferroportin internalization and consequences of a novel ferroportin disease mutation.Am J Hematol. 2017 Oct;92(10):1052-1061. doi: 10.1002/ajh.24844. Epub 2017 Jul 29. Am J Hematol. 2017. PMID: 28681497
-
Iron and hepcidin: a story of recycling and balance.Hematology Am Soc Hematol Educ Program. 2013;2013:1-8. doi: 10.1182/asheducation-2013.1.1. Hematology Am Soc Hematol Educ Program. 2013. PMID: 24319154
Cited by
-
Comparative study of liver injury protection by Akkermansia muciniphila and Faecalibacterium prausnitzii interventions in live and cell-free supernatant forms via targeting the hepcidin - ferroportin axis in mice with CCl₄-induced liver fibrosis.Gut Pathog. 2025 Jul 17;17(1):54. doi: 10.1186/s13099-025-00728-x. Gut Pathog. 2025. PMID: 40676686 Free PMC article.
-
Iron metabolism abnormalities in autoimmune hemolytic anemia and Jianpishengxue keli can ameliorate hemolysis and improve iron metabolism in AIHA mouse models.Ann Med. 2023 Dec;55(1):231-240. doi: 10.1080/07853890.2022.2157475. Ann Med. 2023. PMID: 36576329 Free PMC article.
-
Pre-operative anemia and peri-operative transfusion are associated with poor oncologic outcomes in cancers of the esophagus: potential impact of patient blood management on cancer outcomes.BMC Cancer. 2023 Jan 28;23(1):99. doi: 10.1186/s12885-023-10579-x. BMC Cancer. 2023. PMID: 36709278 Free PMC article.
-
Alteration of osteoclast activity in childhood cancer survivors: Role of iron and of CB2/TRPV1 receptors.PLoS One. 2022 Jul 21;17(7):e0271730. doi: 10.1371/journal.pone.0271730. eCollection 2022. PLoS One. 2022. PMID: 35862357 Free PMC article.
-
Hepcidin mimetics in polycythemia vera: resolving the irony of iron deficiency and erythrocytosis.Curr Opin Hematol. 2023 Mar 1;30(2):45-52. doi: 10.1097/MOH.0000000000000747. Epub 2022 Dec 29. Curr Opin Hematol. 2023. PMID: 36728649 Free PMC article. Review.
References
-
- Babitt JL, Huang FW, Wrighting DM, et al. (2006). Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nature Genetics, 38, 531–539. - PubMed
Further reading
-
- Camaschella C, & Poggiali E (2009). Rare types of genetic hemochromatosis. Acta Haematologica, 122(2–3), 140–145. - PubMed
-
- Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. (1996). A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nature Genetics, 13(4), 399–408. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
