

cAMP-mediated regulation of melanocyte genomic instability: A melanoma-preventive strategy
- PMID: 30798934
- PMCID: PMC7048968
- DOI: 10.1016/bs.apcsb.2018.10.008
cAMP-mediated regulation of melanocyte genomic instability: A melanoma-preventive strategy
Abstract
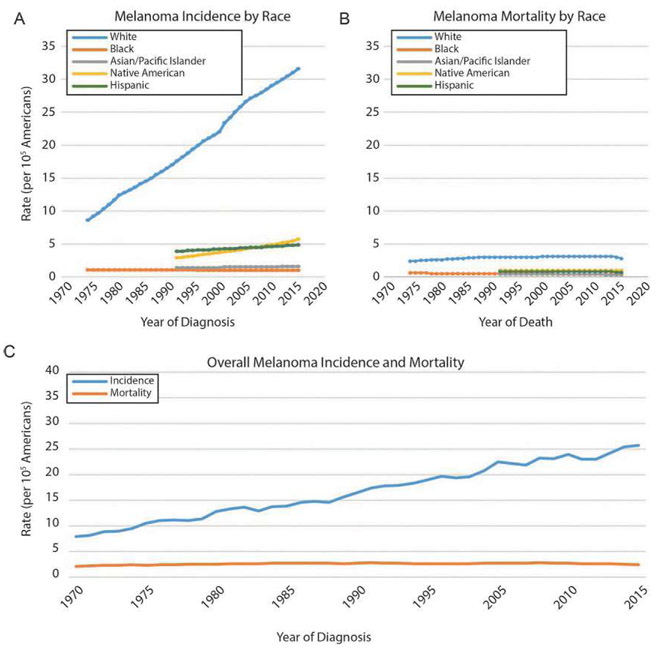
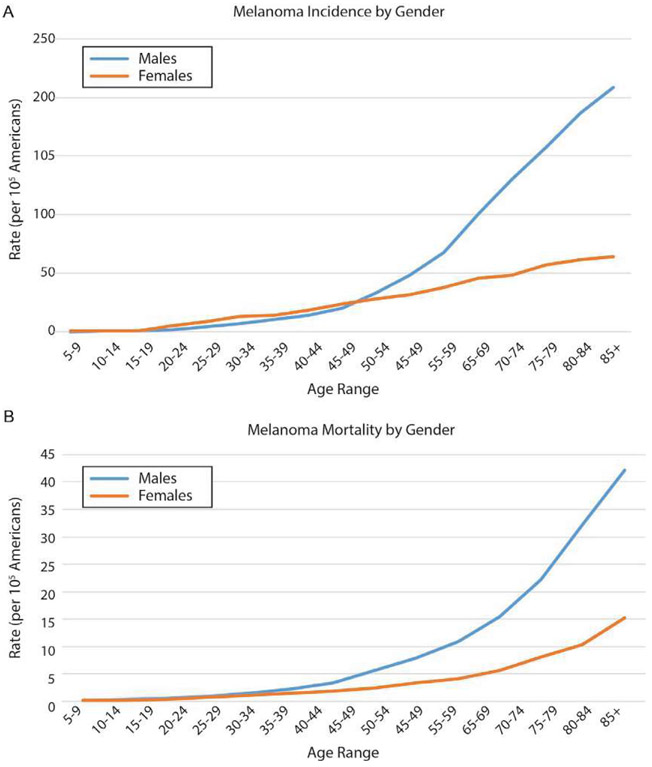
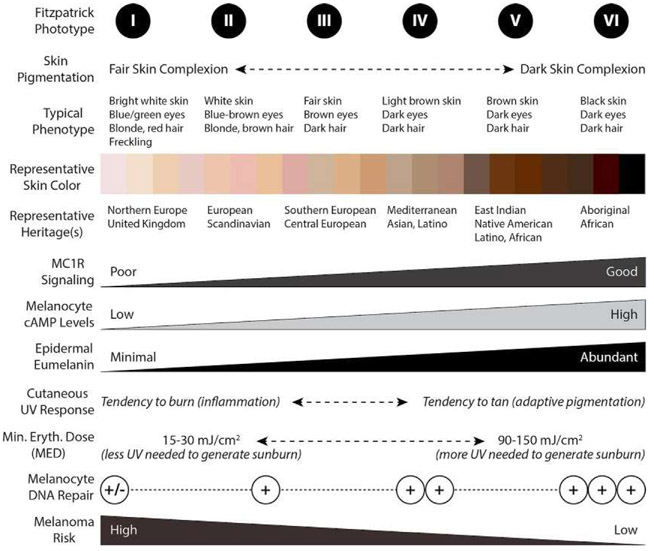
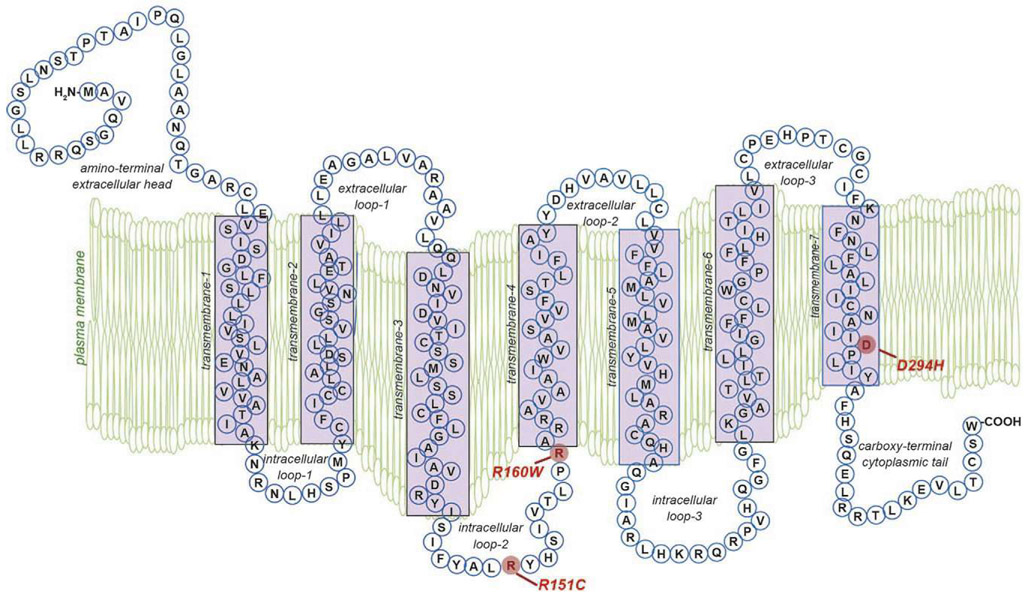
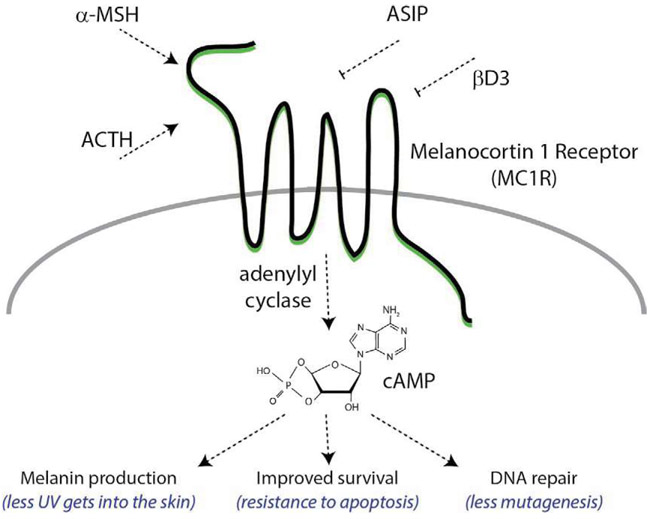
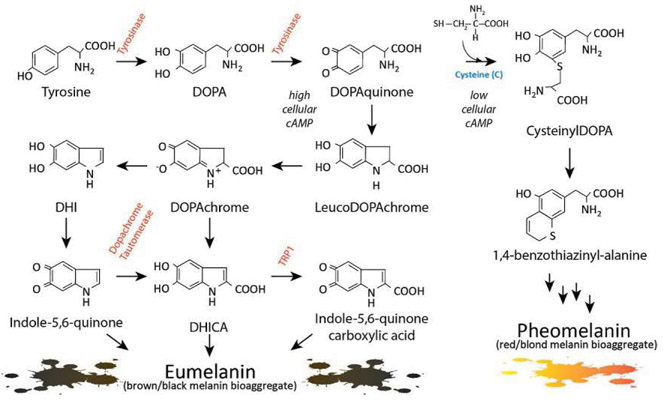
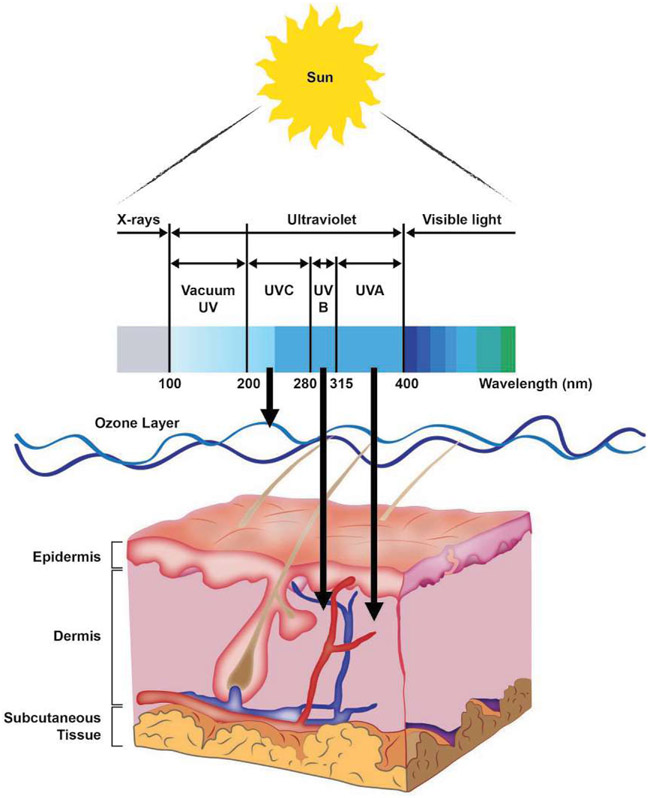
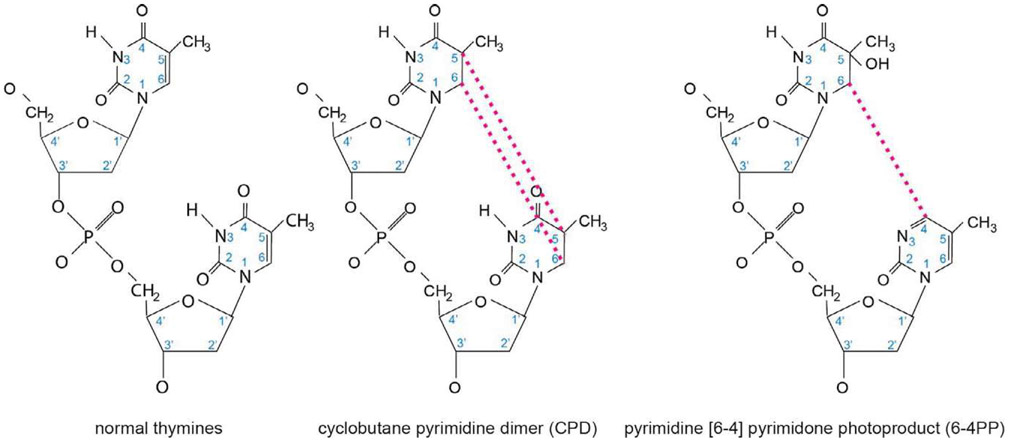
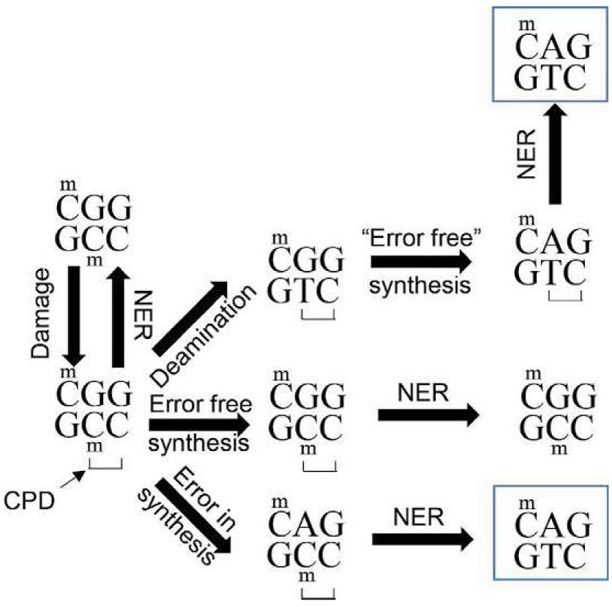
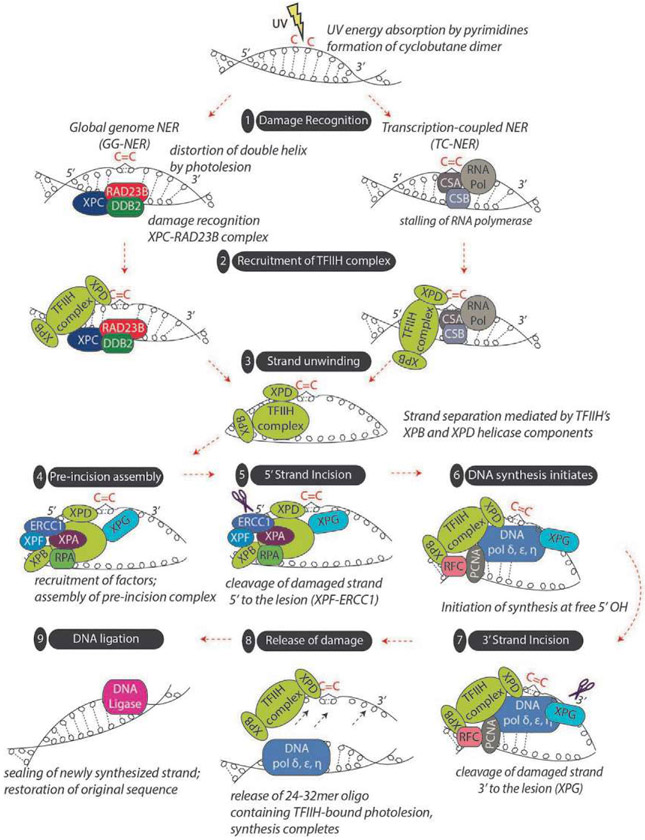
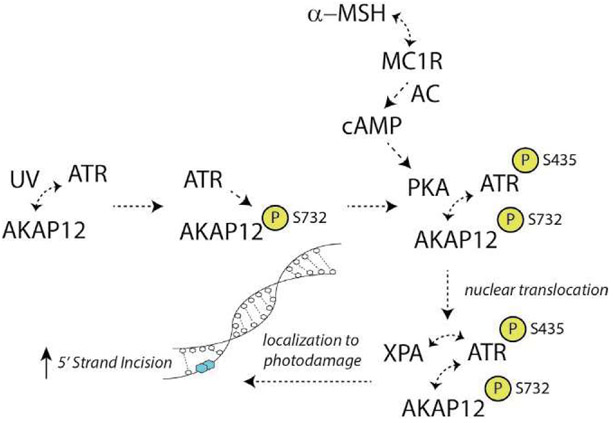
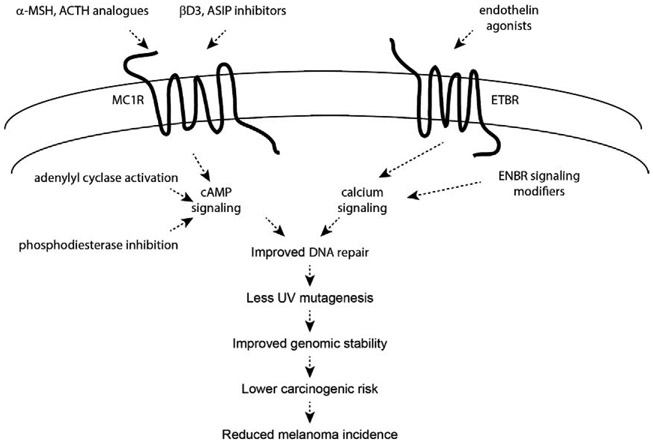
Malignant melanoma of the skin is the leading cause of death from skin cancer and ranks fifth in cancer incidence among all cancers in the United States. While melanoma mortality has remained steady for the past several decades, melanoma incidence has been increasing, particularly among fair-skinned individuals. According to the American Cancer Society, nearly 10,000 people in the United States will die from melanoma this year. Individuals with dark skin complexion are protected damage generated by UV-light due to the high content of UV-blocking melanin pigment in their epidermis as well as better capacity for melanocytes to cope with UV damage. There is now ample evidence that suggests that the melanocortin 1 receptor (MC1R) is a major melanoma risk factor. Inherited loss-of-function mutations in MC1R are common in melanoma-prone persons, correlating with a less melanized skin complexion and poorer recovery from mutagenic photodamage. We and others are interested in the MC1R signaling pathway in melanocytes, its mechanisms of enhancing genomic stability and pharmacologic opportunities to reduce melanoma risk based on those insights. In this chapter, we review melanoma risk factors, the MC1R signaling pathway, and the relationship between MC1R signaling and DNA repair.
Keywords: ATR; MC1R; Melanin; Melanoma; Mutation; Nucleotide excision repair; Risk; UV; cAMP.
© 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflicts of interest
The authors would like to state that there are no conflicts of interest that arose during the preparation of this manuscript.
Figures
References
-
- Abdel-Malek ZA, Ruwe A, Kavanagh-Starner R, Kadekaro AL, Swope V, Haskell-Luevano C, et al. (2009). Alpha-MSH tripeptide analogs activate the melanocortin 1 receptor and reduce UV-induced DNA damage in human melanocytes. Pigment Cell & Melanoma Research, 22(5), 635–644. 10.1111/j.1755-148X.2009.00598.x. - DOI - PubMed
-
- Abdel-Malek ZA, Scott MC, Furumura M, Lamoreux ML, Ollmann M, Barsh GS, et al. (2001). The melanocortin 1 receptor is the principal mediator of the effects of agouti signaling protein on mammalian melanocytes. Journal of Cell Science, 114, 1019–1024 Pt. 5. - PubMed
-
- Abdel-Malek Z, Scott MC, Suzuki I, Tada A, Im S, Lamoreux L, et al. (2000). The melanocortin-1 receptor is a key regulator of human cutaneous pigmentation. Pigment Cell Research, 13(Suppl. 8), 156–162. - PubMed
-
- Abdel-Rahman MH, Pilarski R, Massengill JB, Christopher BN, Noss R, & Davidorf FH (2011). Melanoma candidate genes CDKN2A/p16/INK4A, p14ARF, and CDK4 sequencing in patients with uveal melanoma with relative high-risk for hereditary cancer predisposition. Melanoma Research, 21(3), 175–179. 10.1097/CMR.0b013e328343eca2. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
