

Depletion of the RNA binding protein HNRNPD impairs homologous recombination by inhibiting DNA-end resection and inducing R-loop accumulation
- PMID: 30799487
- PMCID: PMC6486545
- DOI: 10.1093/nar/gkz076
Depletion of the RNA binding protein HNRNPD impairs homologous recombination by inhibiting DNA-end resection and inducing R-loop accumulation
Abstract
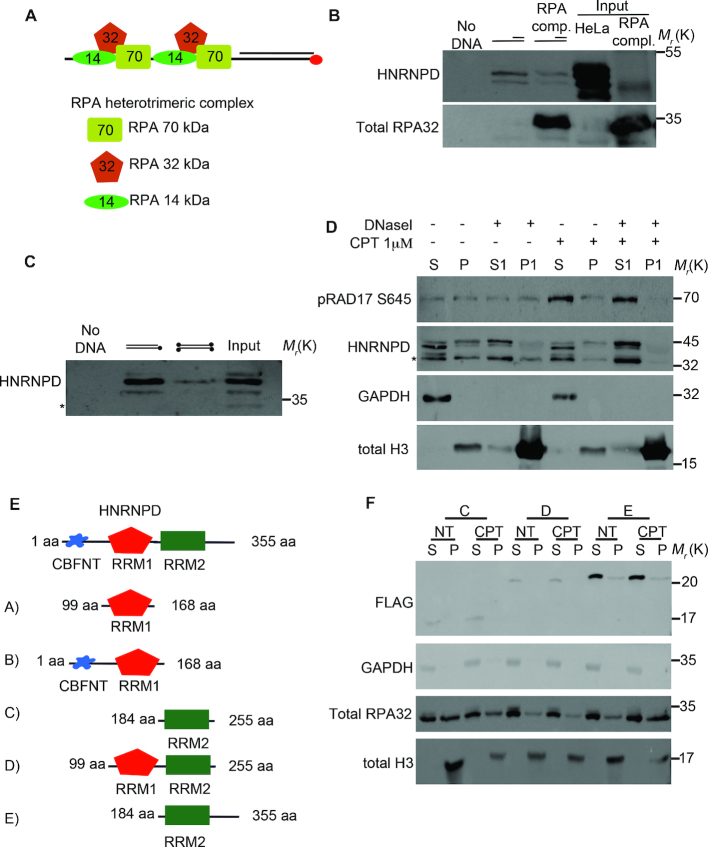
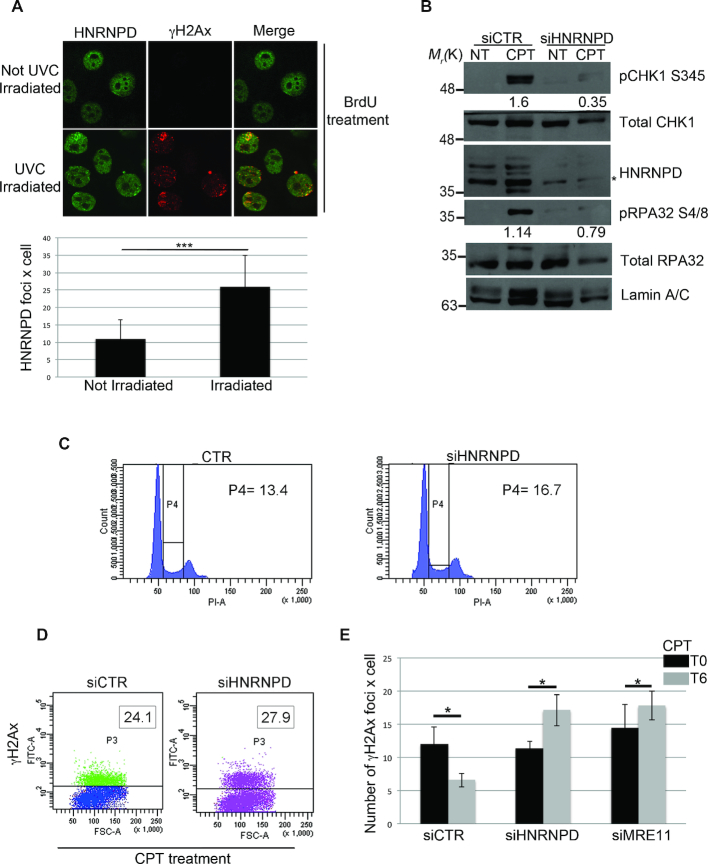
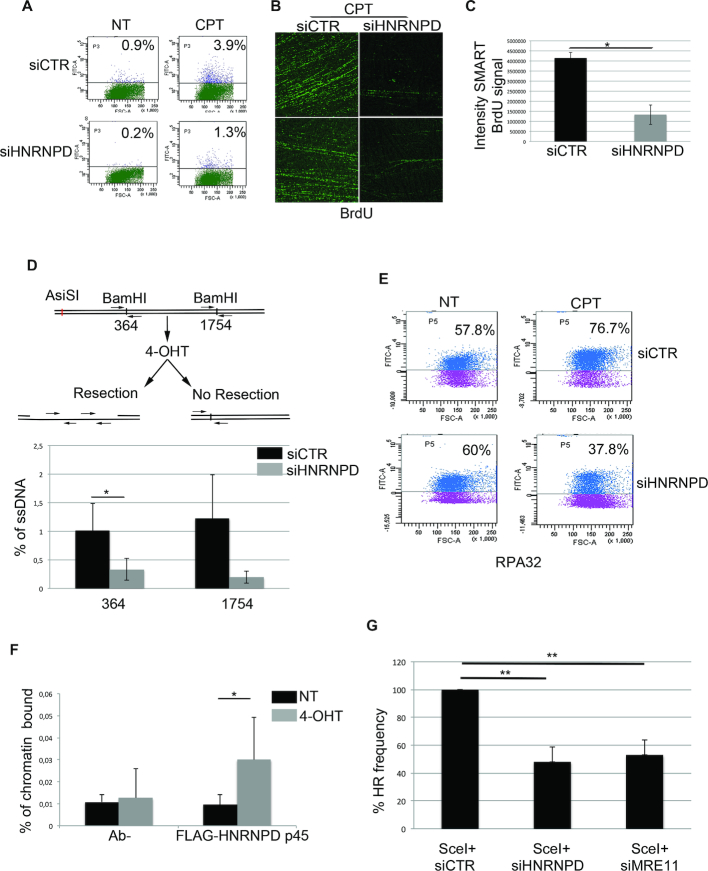
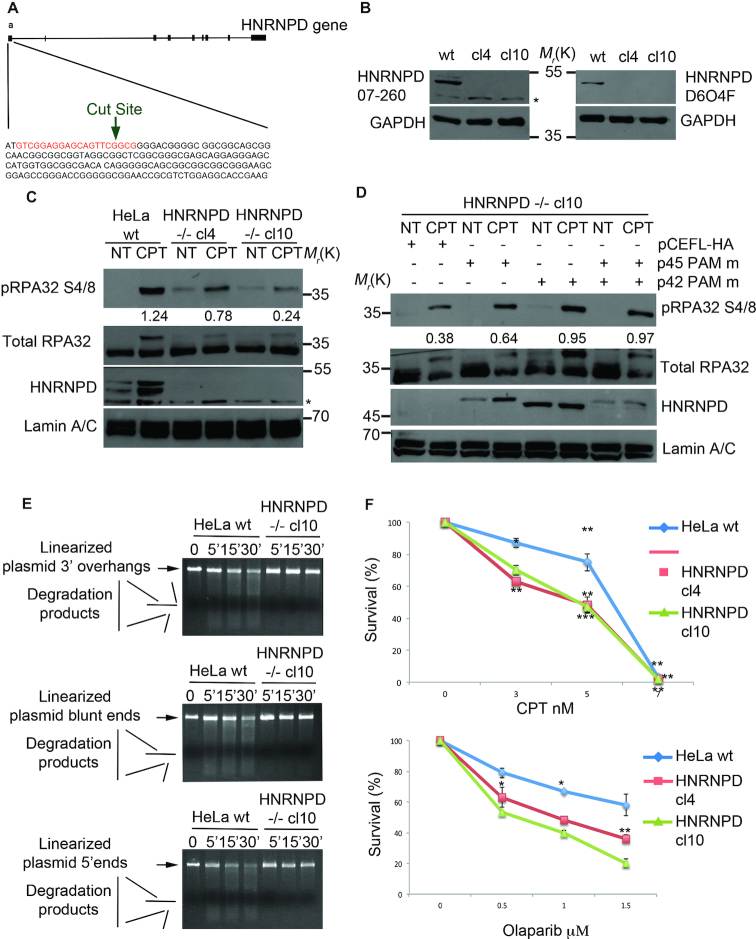
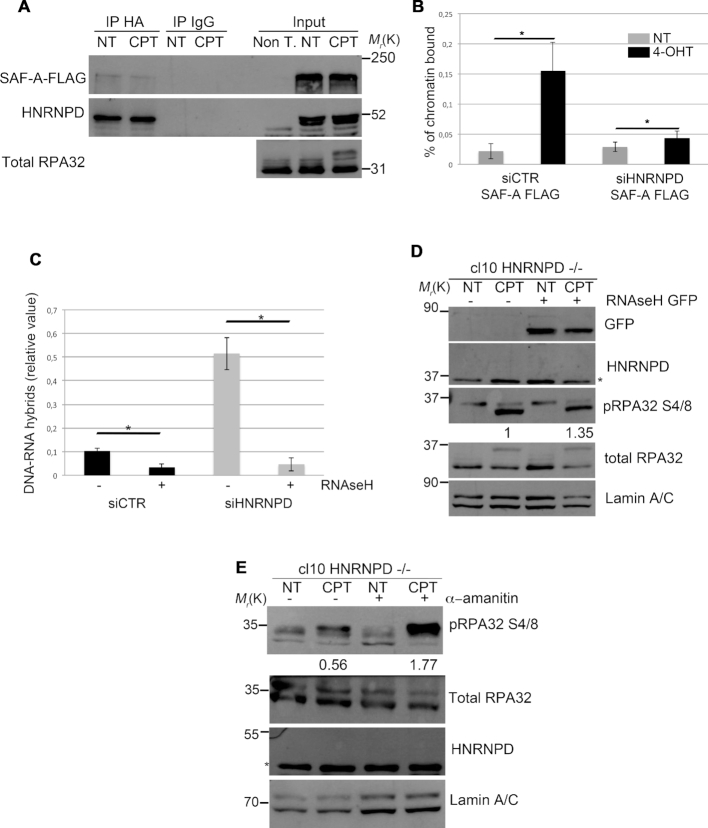
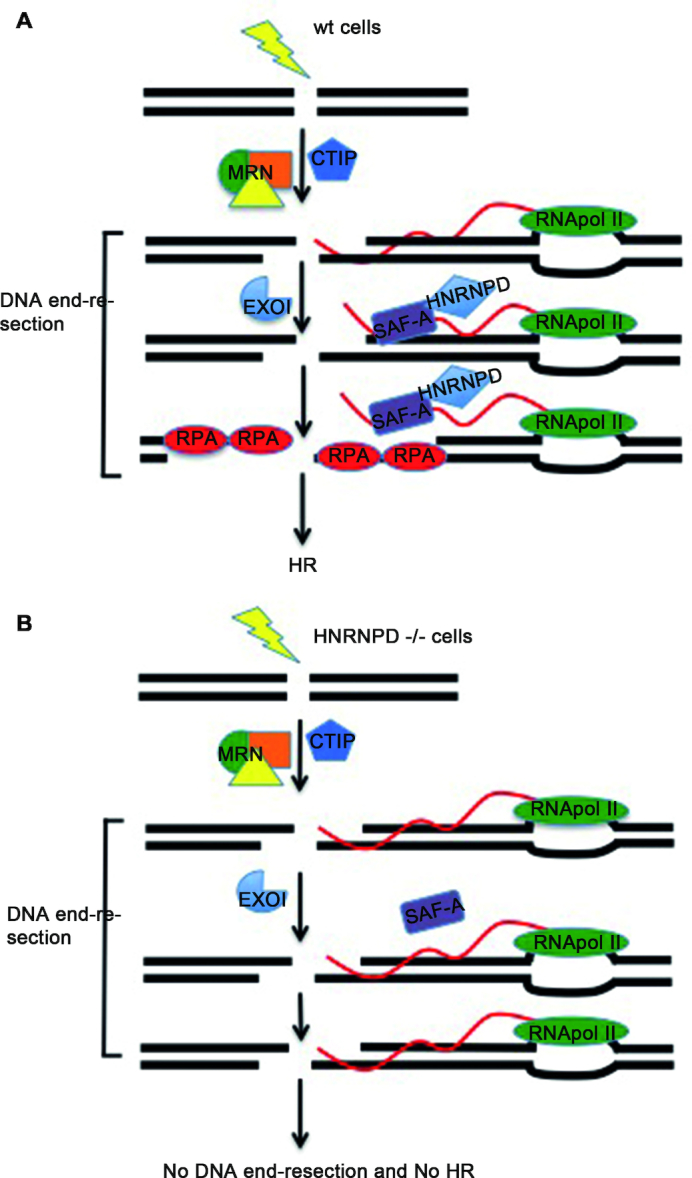
DNA double strand break (DSB) repair through homologous recombination (HR) is crucial to maintain genome stability. DSB resection generates a single strand DNA intermediate, which is crucial for the HR process. We used a synthetic DNA structure, mimicking a resection intermediate, as a bait to identify proteins involved in this process. Among these, LC/MS analysis identified the RNA binding protein, HNRNPD. We found that HNRNPD binds chromatin, although this binding occurred independently of DNA damage. However, upon damage, HNRNPD re-localized to γH2Ax foci and its silencing impaired CHK1 S345 phosphorylation and the DNA end resection process. Indeed, HNRNPD silencing reduced: the ssDNA fraction upon camptothecin treatment; AsiSI-induced DSB resection; and RPA32 S4/8 phosphorylation. CRISPR/Cas9-mediated HNRNPD knockout impaired in vitro DNA resection and sensitized cells to camptothecin and olaparib treatment. We found that HNRNPD interacts with the heterogeneous nuclear ribonucleoprotein SAF-A previously associated with DNA damage repair. HNRNPD depletion resulted in an increased amount of RNA:DNA hybrids upon DNA damage. Both the expression of RNase H1 and RNA pol II inhibition recovered the ability to phosphorylate RPA32 S4/8 in HNRNPD knockout cells upon DNA damage, suggesting that RNA:DNA hybrid resolution likely rescues the defective DNA damage response of HNRNPD-depleted cells.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
