

Challenges Facing Airway Epithelial Cell-Based Therapy for Cystic Fibrosis
- PMID: 30800069
- PMCID: PMC6376457
- DOI: 10.3389/fphar.2019.00074
Challenges Facing Airway Epithelial Cell-Based Therapy for Cystic Fibrosis
Abstract
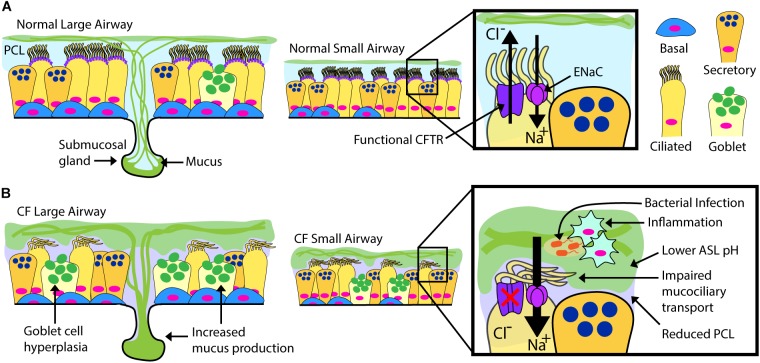
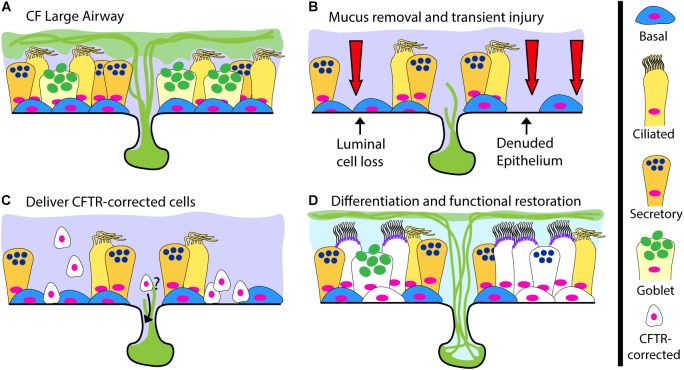
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene cause the life-limiting hereditary disease, cystic fibrosis (CF). Decreased or absent functional CFTR protein in airway epithelial cells leads to abnormally viscous mucus and impaired mucociliary transport, resulting in bacterial infections and inflammation causing progressive lung damage. There are more than 2000 known variants in the CFTR gene. A subset of CF individuals with specific CFTR mutations qualify for pharmacotherapies of variable efficacy. These drugs, termed CFTR modulators, address key defects in protein folding, trafficking, abundance, and function at the apical cell membrane resulting from specific CFTR mutations. However, some CFTR mutations result in little or no CFTR mRNA or protein expression for which a pharmaceutical strategy is more challenging and remote. One approach to rescue CFTR function in the airway epithelium is to replace cells that carry a mutant CFTR sequence with cells that express a normal copy of the gene. Cell-based therapy theoretically has the potential to serve as a one-time cure for CF lung disease regardless of the causative CFTR mutation. In this review, we explore major challenges and recent progress toward this ambitious goal. The ideal therapeutic cell would: (1) be autologous to avoid the complications of rejection and immune-suppression; (2) be safely modified to express functional CFTR; (3) be expandable ex vivo to generate sufficient cell quantities to restore CFTR function; and (4) have the capacity to engraft, proliferate and persist long-term in recipient airways without complications. Herein, we explore human bronchial epithelial cells (HBECs) and induced pluripotent stem cells (iPSCs) as candidate cell therapies for CF and explore the challenges facing their delivery to the human airway.
Keywords: cell-based therapy; cystic fibrosis; engraftment; human bronchial epithelial cells; induced pluripotent stem cells.
Figures
References
-
- Bove P. F., Randell S. H. (2015). “Comparative mammalian lung primary surface epithelial cell culture,” in Comparative Biology of the Normal Lung, (Cambridge, MA: Academic Press; ), 129–139. 10.1016/B978-0-12-404577-4.00011-4 - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources