

Somatic mutations in early metazoan genes disrupt regulatory links between unicellular and multicellular genes in cancer
- PMID: 30803482
- PMCID: PMC6402835
- DOI: 10.7554/eLife.40947
Somatic mutations in early metazoan genes disrupt regulatory links between unicellular and multicellular genes in cancer
Abstract
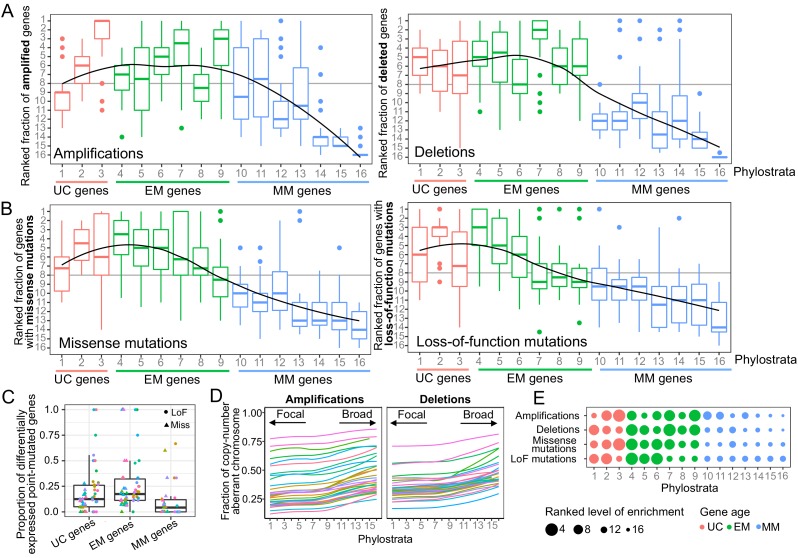
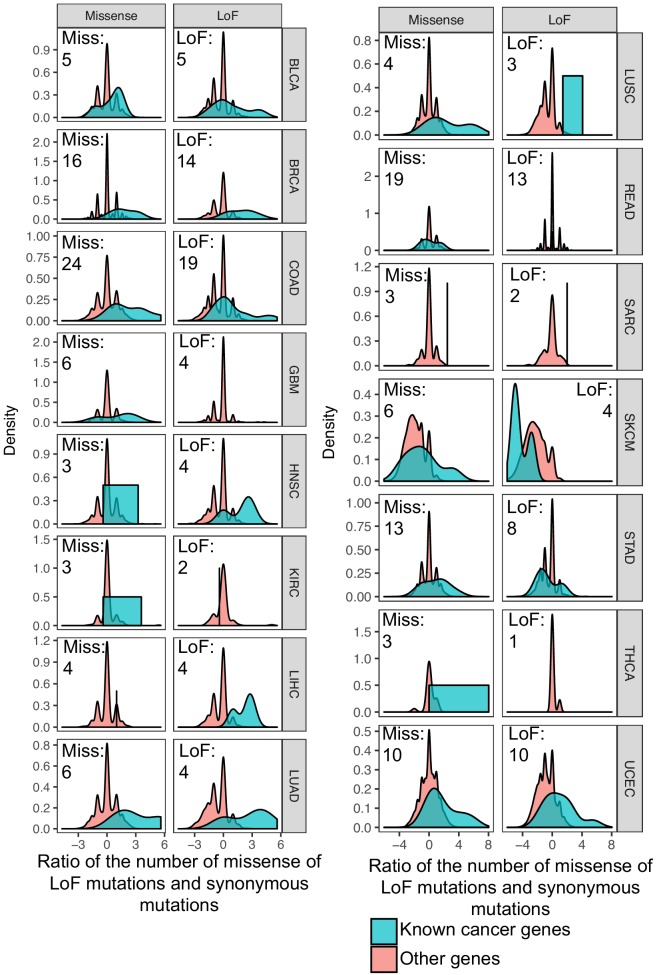
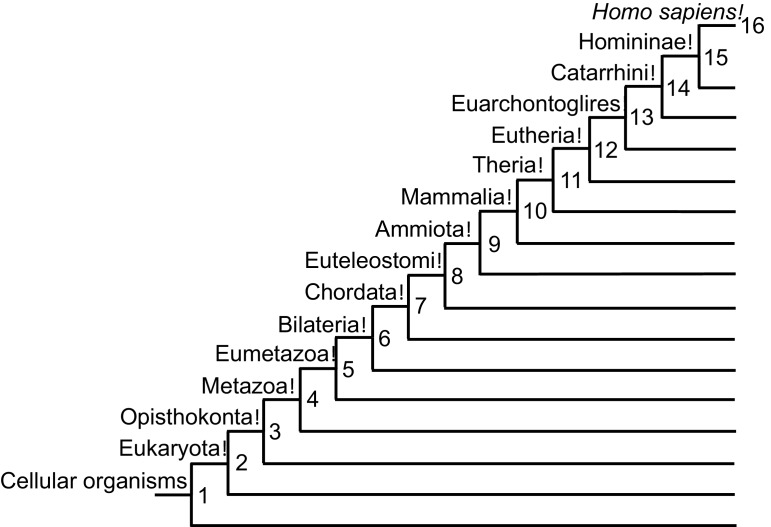
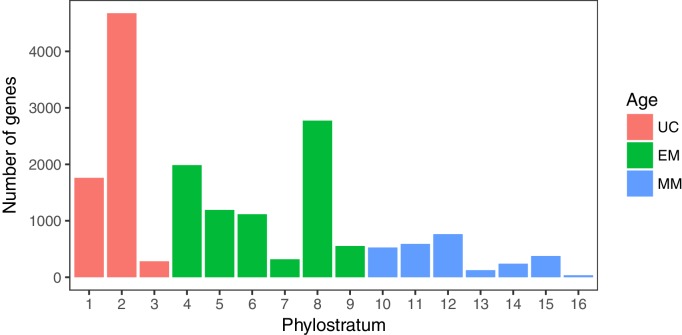
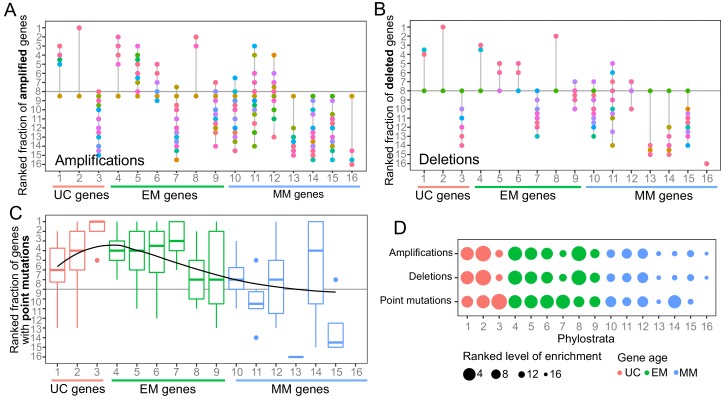
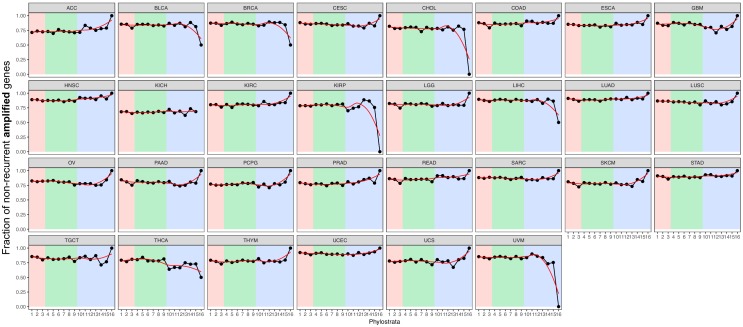
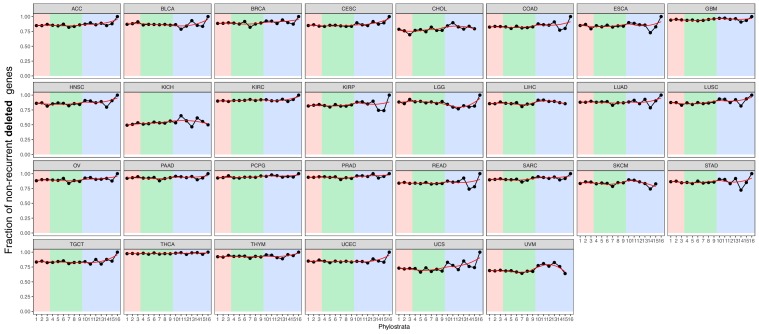
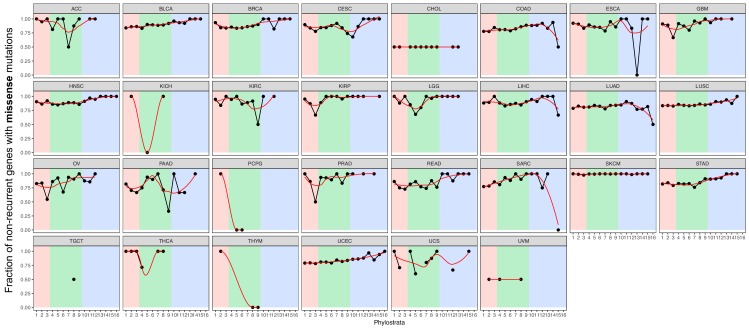
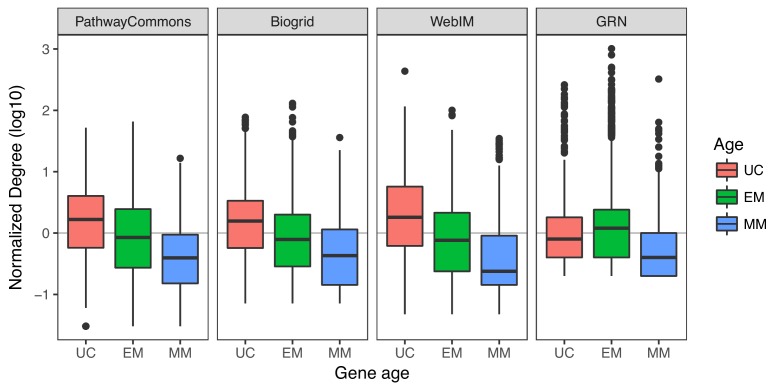
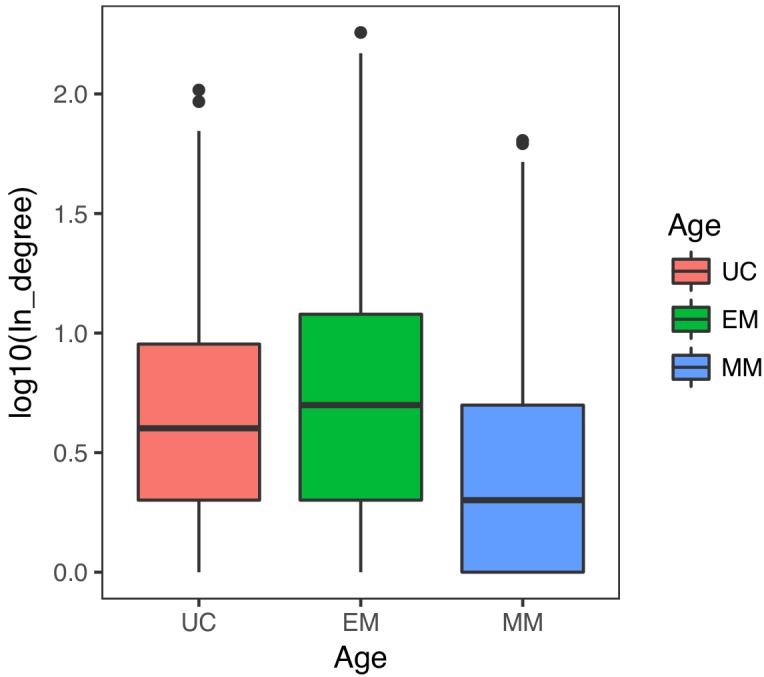
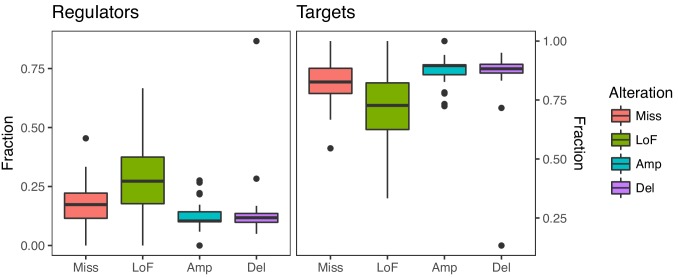
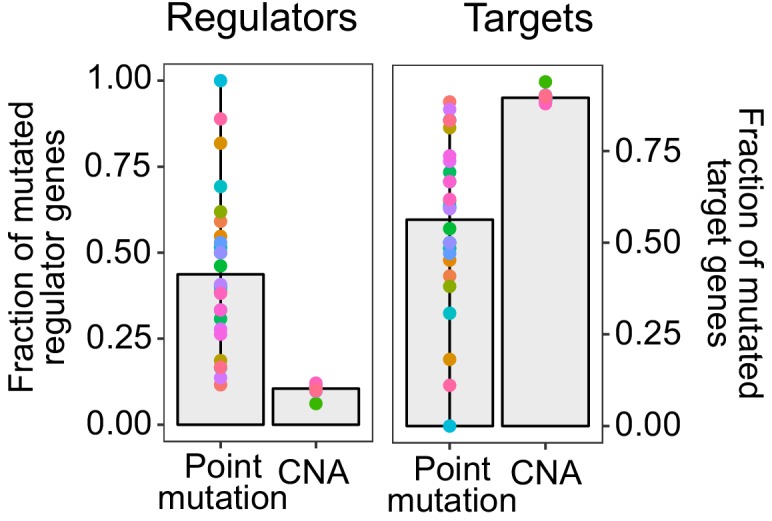
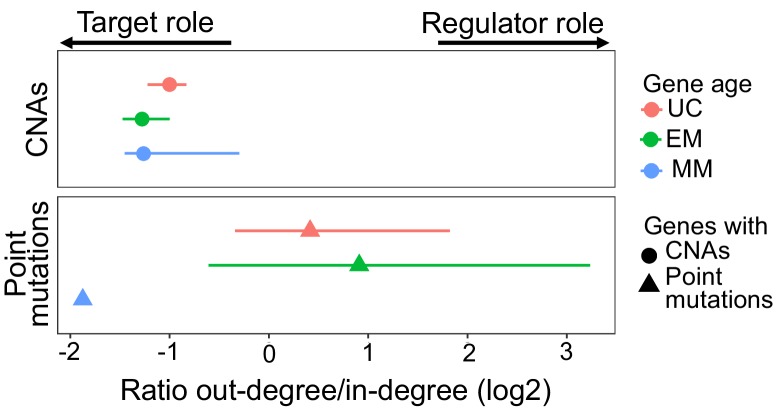
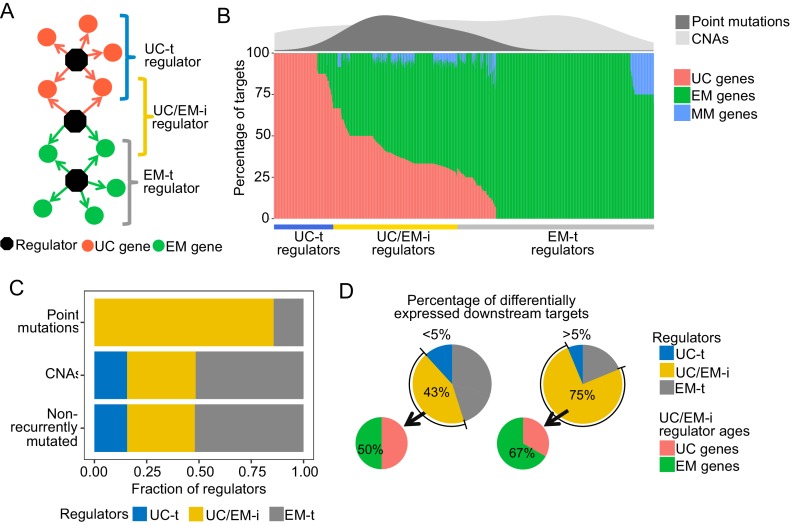
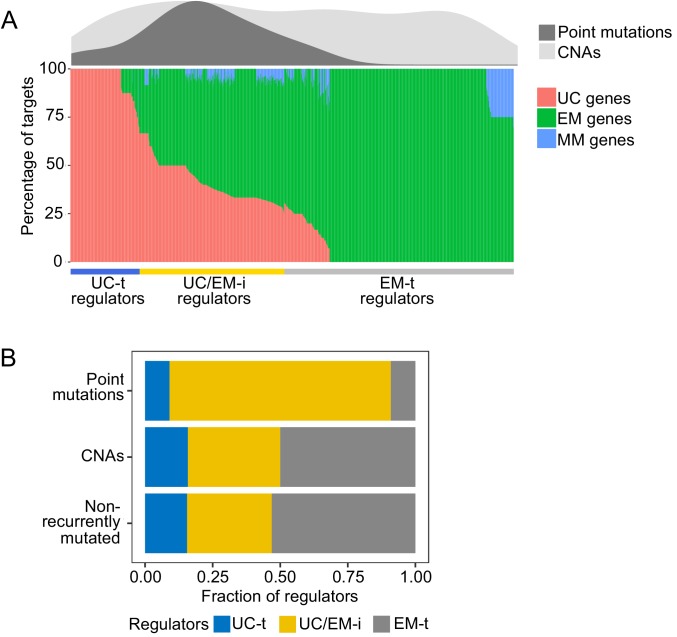
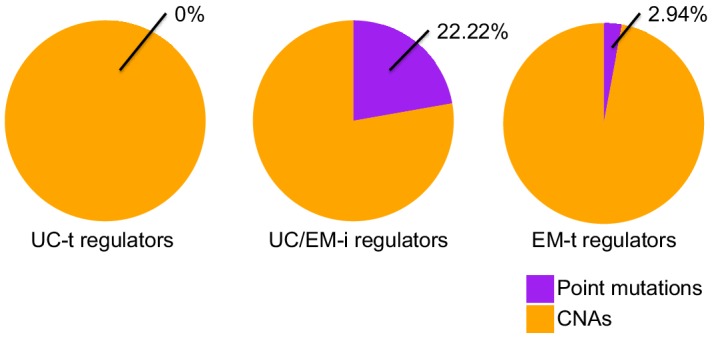
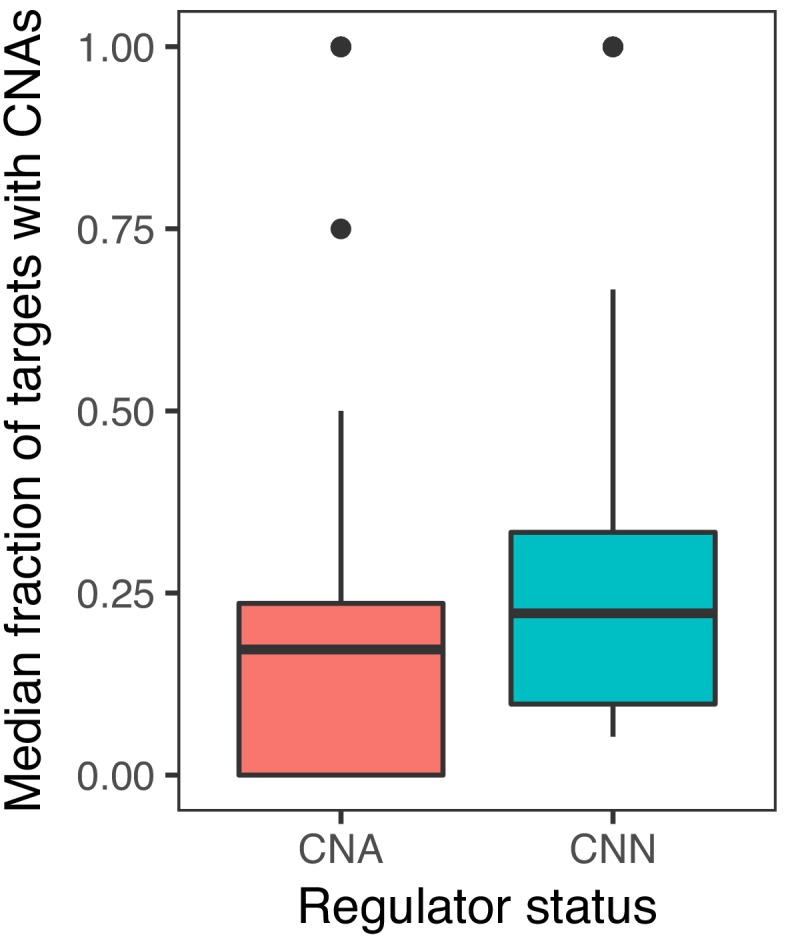
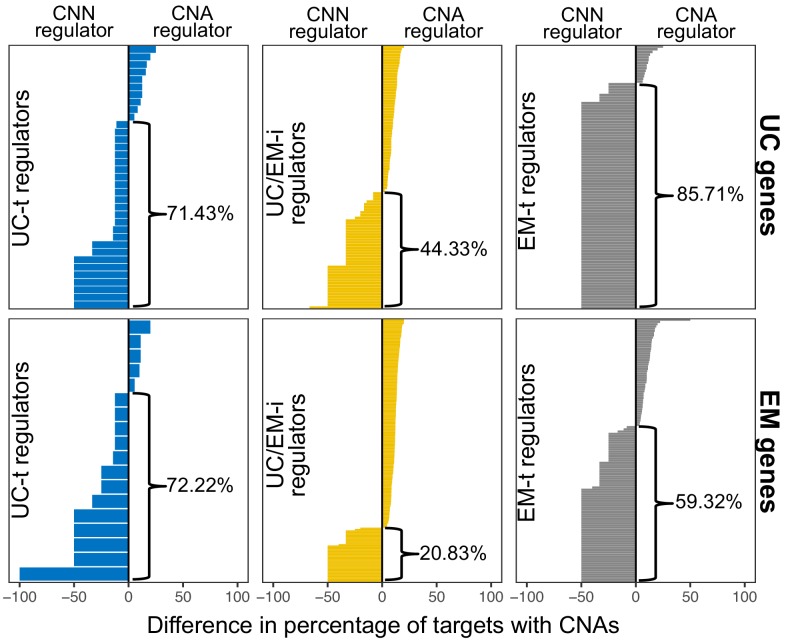
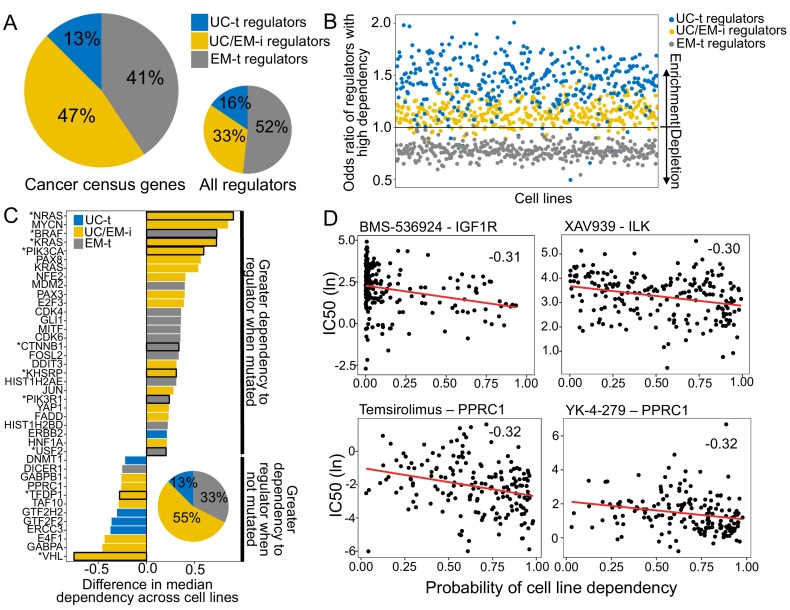
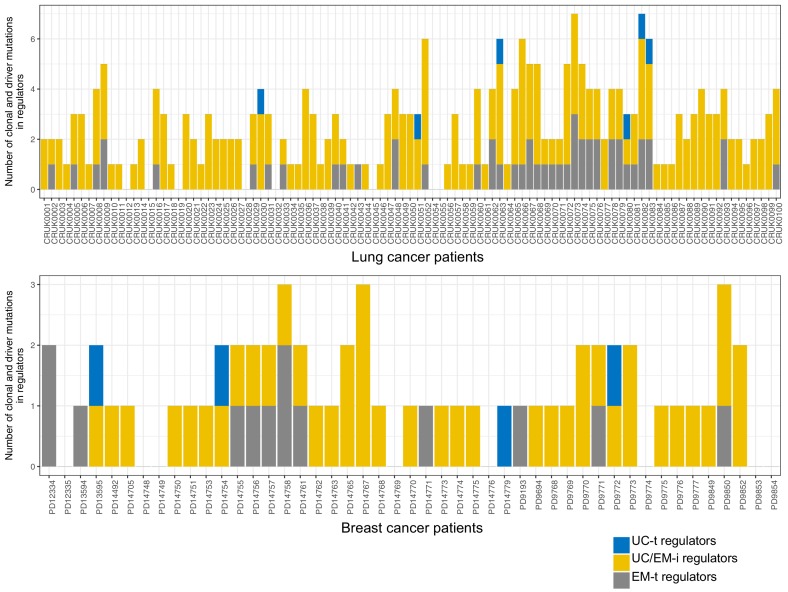
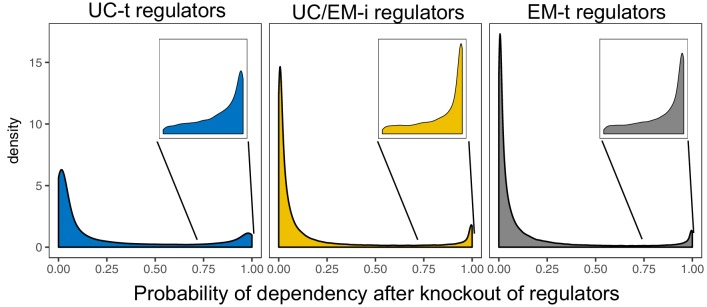
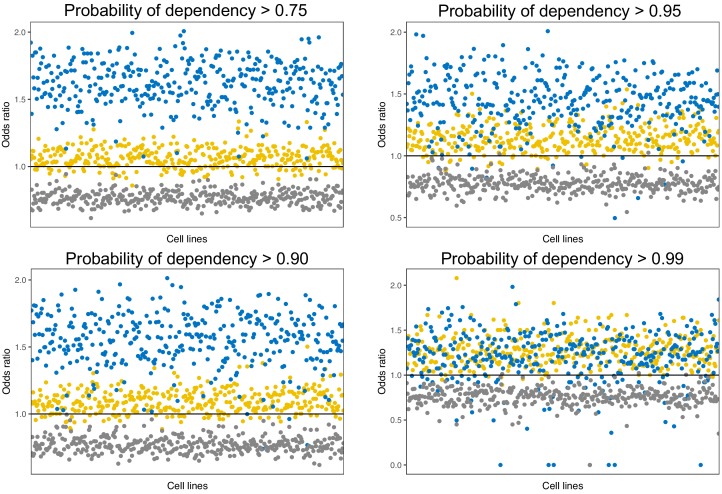
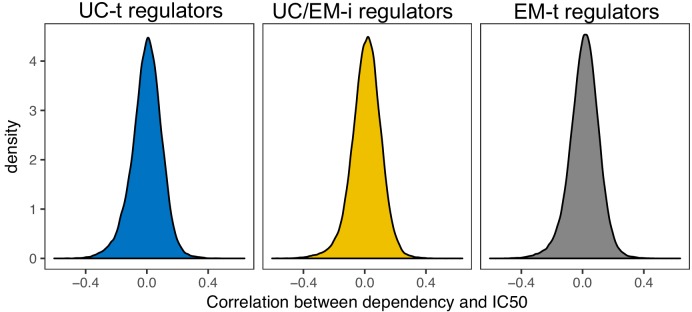
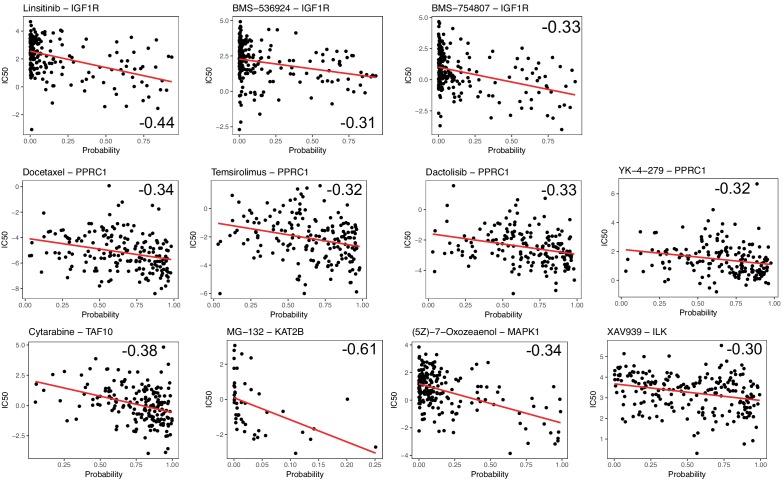
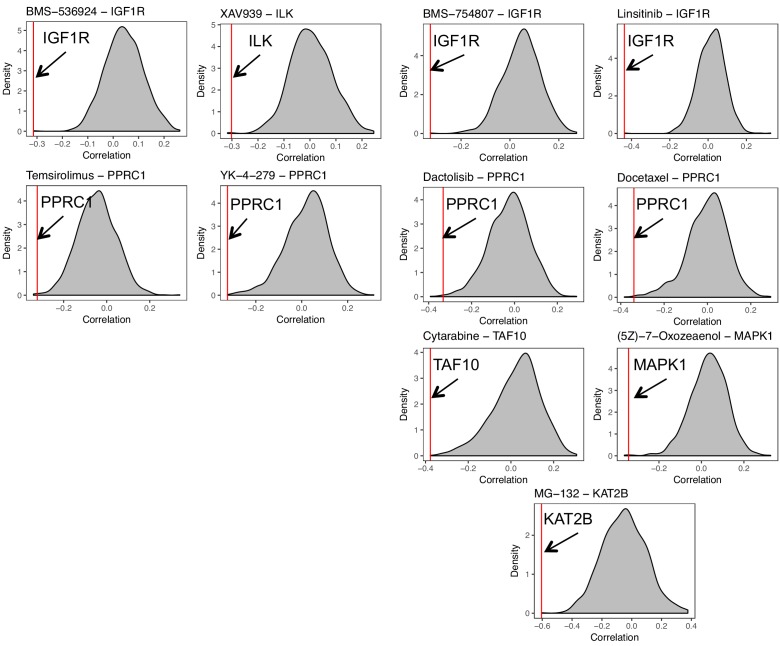
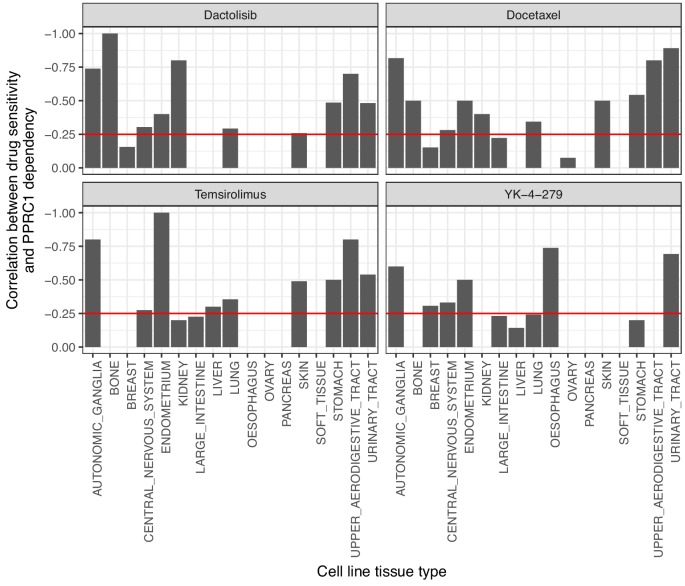
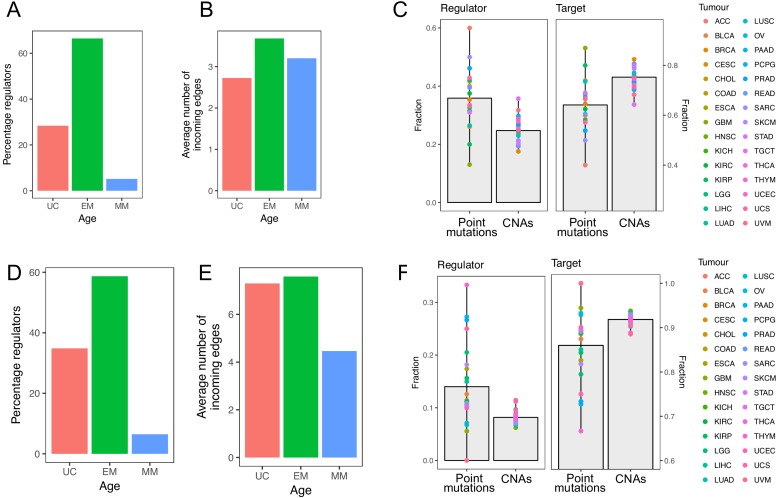
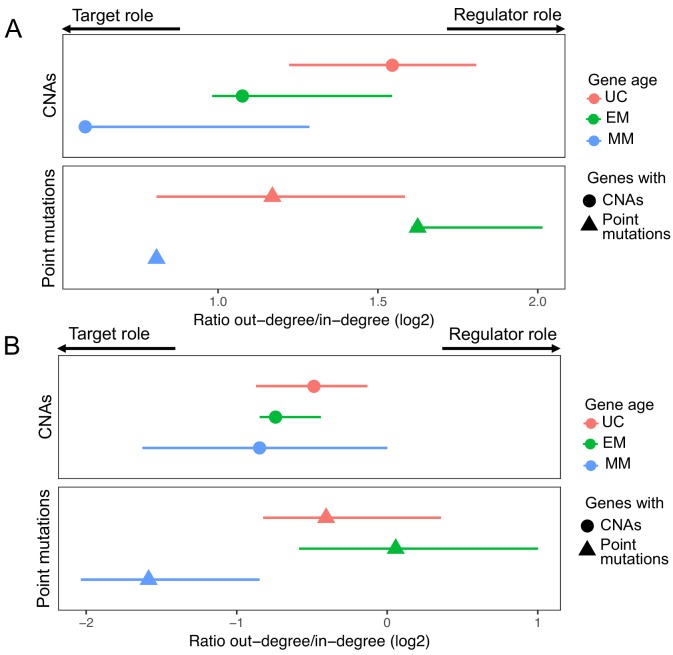
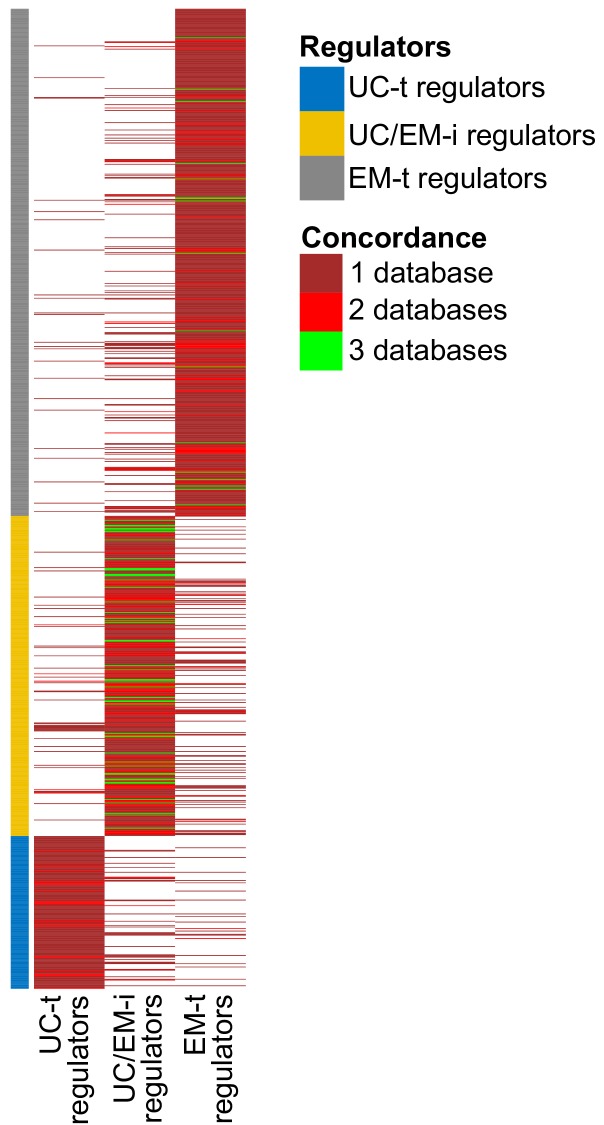
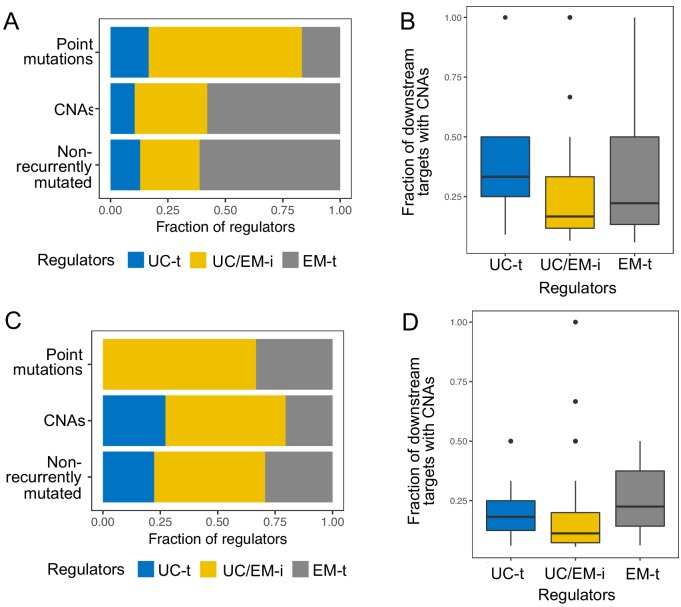
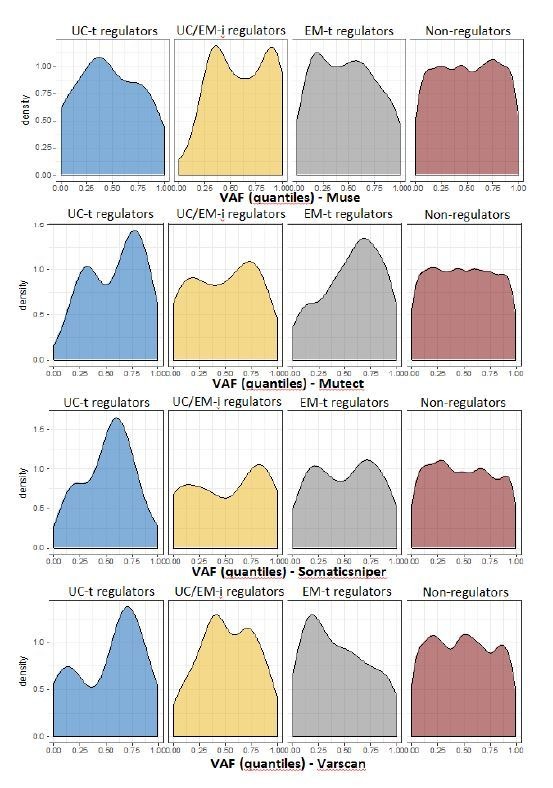
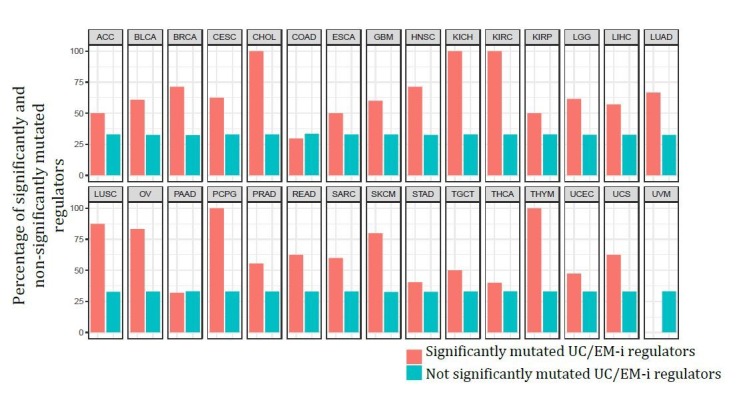
Extensive transcriptional alterations are observed in cancer, many of which activate core biological processes established in unicellular organisms or suppress differentiation pathways formed in metazoans. Through rigorous, integrative analysis of genomics data from a range of solid tumors, we show many transcriptional changes in tumors are tied to mutations disrupting regulatory interactions between unicellular and multicellular genes within human gene regulatory networks (GRNs). Recurrent point mutations were enriched in regulator genes linking unicellular and multicellular subnetworks, while copy-number alterations affected downstream target genes in distinctly unicellular and multicellular regions of the GRN. Our results depict drivers of tumourigenesis as genes that created key regulatory links during the evolution of early multicellular life, whose dysfunction creates widespread dysregulation of primitive elements of the GRN. Several genes we identified as important in this process were associated with drug response, demonstrating the potential clinical value of our approach.
Keywords: cancer; cancer biology; computational biology; evolution; human; multicellularity; mutations; networks; orthology; systems biology.
© 2019, Trigos et al.
Conflict of interest statement
AT, RP, AP, DG No competing interests declared
Figures
Similar articles
-
Disruption of metazoan gene regulatory networks in cancer alters the balance of co-expression between genes of unicellular and multicellular origins.Genome Biol. 2024 Apr 29;25(1):110. doi: 10.1186/s13059-024-03247-1. Genome Biol. 2024. PMID: 38685127 Free PMC article.
-
Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors.Proc Natl Acad Sci U S A. 2017 Jun 13;114(24):6406-6411. doi: 10.1073/pnas.1617743114. Epub 2017 May 8. Proc Natl Acad Sci U S A. 2017. PMID: 28484005 Free PMC article.
-
How the evolution of multicellularity set the stage for cancer.Br J Cancer. 2018 Jan;118(2):145-152. doi: 10.1038/bjc.2017.398. Epub 2018 Jan 16. Br J Cancer. 2018. PMID: 29337961 Free PMC article. Review.
-
Urothelial cancer gene regulatory networks inferred from large-scale RNAseq, Bead and Oligo gene expression data.BMC Syst Biol. 2015 May 14;9:21. doi: 10.1186/s12918-015-0165-z. BMC Syst Biol. 2015. PMID: 25971253 Free PMC article.
-
aCLS cancers: Genomic and epigenetic changes transform the cell of origin of cancer into a tumorigenic pathogen of unicellular organization and lifestyle.Gene. 2020 Feb 5;726:144174. doi: 10.1016/j.gene.2019.144174. Epub 2019 Oct 21. Gene. 2020. PMID: 31647999 Review.
Cited by
-
Cancer genes and cancer stem cells in tumorigenesis: Evolutionary deep homology and controversies.Genes Dis. 2022 Apr 4;9(5):1234-1247. doi: 10.1016/j.gendis.2022.03.010. eCollection 2022 Sep. Genes Dis. 2022. PMID: 35873035 Free PMC article. Review.
-
Reverting to single-cell biology: The predictions of the atavism theory of cancer.Prog Biophys Mol Biol. 2021 Oct;165:49-55. doi: 10.1016/j.pbiomolbio.2021.08.002. Epub 2021 Aug 8. Prog Biophys Mol Biol. 2021. PMID: 34371024 Free PMC article.
-
Global chromatin reorganization and regulation of genes with specific evolutionary ages during differentiation and cancer.bioRxiv [Preprint]. 2024 Oct 14:2023.10.30.564438. doi: 10.1101/2023.10.30.564438. bioRxiv. 2024. Update in: Nucleic Acids Res. 2025 Feb 08;53(4):gkaf084. doi: 10.1093/nar/gkaf084. PMID: 39149250 Free PMC article. Updated. Preprint.
-
On the gene expression landscape of cancer.PLoS One. 2023 Feb 21;18(2):e0277786. doi: 10.1371/journal.pone.0277786. eCollection 2023. PLoS One. 2023. PMID: 36802377 Free PMC article.
-
Disruption of metazoan gene regulatory networks in cancer alters the balance of co-expression between genes of unicellular and multicellular origins.Genome Biol. 2024 Apr 29;25(1):110. doi: 10.1186/s13059-024-03247-1. Genome Biol. 2024. PMID: 38685127 Free PMC article.
References
-
- Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, Chatila WK, Chakravarty D, Han GC, Coleman I, Montgomery B, Pritchard C, Morrissey C, Barbieri CE, Beltran H, Sboner A, Zafeiriou Z, Miranda S, Bielski CM, Penson AV, Tolonen C, Huang FW, Robinson D, Wu YM, Lonigro R, Garraway LA, Demichelis F, Kantoff PW, Taplin ME, Abida W, Taylor BS, Scher HI, Nelson PS, de Bono JS, Rubin MA, Sawyers CL, Chinnaiyan AM, Schultz N, Van Allen EM, PCF/SU2C International Prostate Cancer Dream Team The long tail of oncogenic drivers in prostate cancer. Nature Genetics. 2018;50:645–651. doi: 10.1038/s41588-018-0078-z. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
