

Decreased mRNA and protein stability of W1282X limits response to modulator therapy
- PMID: 30803905
- PMCID: PMC6706327
- DOI: 10.1016/j.jcf.2019.02.009
Decreased mRNA and protein stability of W1282X limits response to modulator therapy
Abstract
Background: Cell-based studies have shown that W1282X generates a truncated protein that can be functionally augmented by modulators. However, modulator treatment of primary cells from individuals who carry two copies of W1282X generates no functional CFTR. To understand the lack of response to modulators, we investigated the effect of W1282X on CFTR RNA transcript levels.
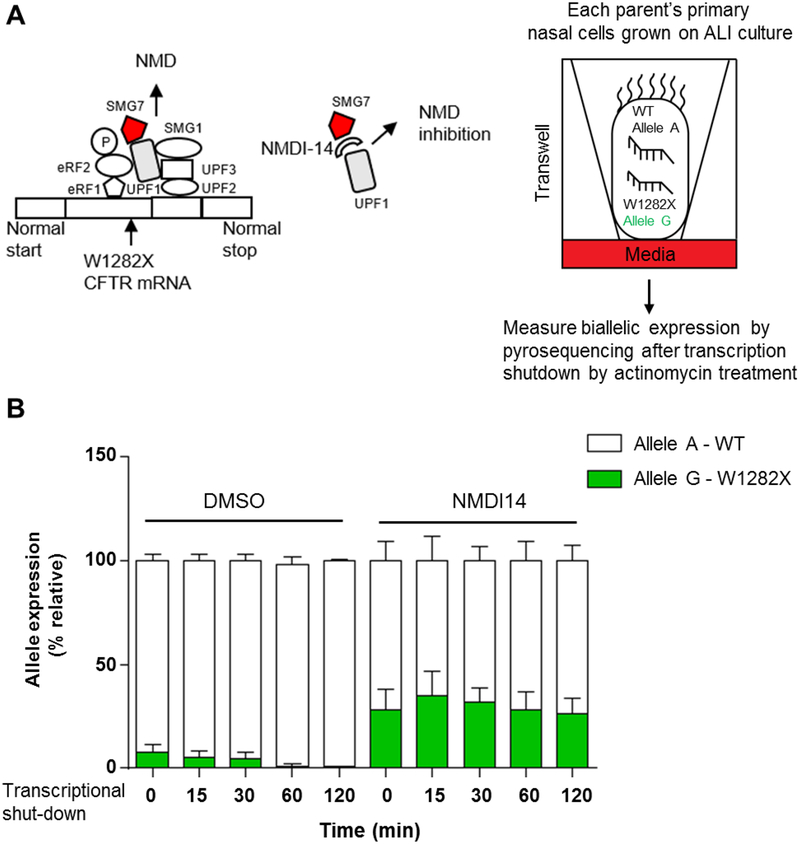
Methods: qRT-PCR and RNA-seq were performed on primary nasal epithelial (NE) cells of a previously studied individual who is homozygous for W1282X, her carrier parents and control individuals without nonsense variants in CFTR.
Results: CFTR RNA bearing W1282X in NE cells shows a steady-state level of 4.2 ± 0.9% of wild-type (WT) CFTR RNA in the mother and 12.4 ± 1.3% in the father. NMDI14, an inhibitor of nonsense-mediated mRNA decay (NMD), restored W1282X mRNA to almost 50% of WT levels in the parental NE cells. RNA-seq of the NE cells homozygous for W1282X showed that CFTR transcript level was reduced to 1.7% of WT (p-value: 4.6e-3). Negligible truncated CFTR protein was generated by Flp-In 293 cells stably expressing the W1282X EMG even though CFTR transcript was well above levels observed in the parents and proband. Finally, we demonstrated that NMD inhibition improved the stability and response to correctors of W1282X-CFTR protein expressed in the Flp-In-293 cells.
Conclusion: These results show that W1282X can cause substantial degradation of CFTR mRNA that has to be addressed before efforts aimed at augmenting CFTR protein function can be effective.
Copyright © 2019 European Cystic Fibrosis Society. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Conflict of Interest
All authors declare that we have no personal, professional or financial conflict of interest.
Figures



Similar articles
-
Functional rescue of c.3846G>A (W1282X) in patient-derived nasal cultures achieved by inhibition of nonsense mediated decay and protein modulators with complementary mechanisms of action.J Cyst Fibros. 2020 Sep;19(5):717-727. doi: 10.1016/j.jcf.2019.12.001. Epub 2019 Dec 9. J Cyst Fibros. 2020. PMID: 31831337
-
Nonsense-mediated RNA Decay Pathway Inhibition Restores Expression and Function of W1282X CFTR.Am J Respir Cell Mol Biol. 2019 Sep;61(3):290-300. doi: 10.1165/rcmb.2018-0316OC. Am J Respir Cell Mol Biol. 2019. PMID: 30836009
-
Use of adenine base editing and homology-independent targeted integration strategies to correct the cystic fibrosis causing variant, W1282X.Hum Mol Genet. 2023 Nov 17;32(23):3237-3248. doi: 10.1093/hmg/ddad143. Hum Mol Genet. 2023. PMID: 37649273 Free PMC article.
-
Pharmacologic therapy for stop mutations: how much CFTR activity is enough?Curr Opin Pulm Med. 2004 Nov;10(6):547-52. doi: 10.1097/01.mcp.0000141247.22078.46. Curr Opin Pulm Med. 2004. PMID: 15510065 Review.
-
Pharmacological induction of CFTR function in patients with cystic fibrosis: mutation-specific therapy.Pediatr Pulmonol. 2005 Sep;40(3):183-96. doi: 10.1002/ppul.20200. Pediatr Pulmonol. 2005. PMID: 15880796 Review.
Cited by
-
Nanomolar-potency 'co-potentiator' therapy for cystic fibrosis caused by a defined subset of minimal function CFTR mutants.Sci Rep. 2019 Nov 27;9(1):17640. doi: 10.1038/s41598-019-54158-2. Sci Rep. 2019. PMID: 31776420 Free PMC article.
-
L1077P CFTR pathogenic variant function rescue by Elexacaftor-Tezacaftor-Ivacaftor in cystic fibrosis patient-derived air-liquid interface (ALI) cultures and organoids: in vitro guided personalized therapy of non-F508del patients.Respir Res. 2023 Sep 6;24(1):217. doi: 10.1186/s12931-023-02516-0. Respir Res. 2023. PMID: 37674160 Free PMC article.
-
Racial inequities and rare CFTR variants: Impact on cystic fibrosis diagnosis and treatment.J Clin Transl Endocrinol. 2024 Apr 20;36:100344. doi: 10.1016/j.jcte.2024.100344. eCollection 2024 Jun. J Clin Transl Endocrinol. 2024. PMID: 38765466 Free PMC article.
-
A W1282X cystic fibrosis mouse allows the study of pharmacological and gene-editing therapeutics to restore CFTR function.J Cyst Fibros. 2025 Jan;24(1):164-174. doi: 10.1016/j.jcf.2024.10.008. Epub 2024 Nov 12. J Cyst Fibros. 2025. PMID: 39532588
-
Site-Specific RNA Editing of Stop Mutations in the CFTR mRNA of Human Bronchial Cultured Cells.Int J Mol Sci. 2023 Jun 30;24(13):10940. doi: 10.3390/ijms241310940. Int J Mol Sci. 2023. PMID: 37446121 Free PMC article.
References
-
- Wainwright CE, Elborn JS, Ramsey BW. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(18):1783–4. - PubMed
-
- Taylor-Cousar JL, Munck A, McKone EF, van der Ent CK, Moeller A, Simard C, et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med. 2017;377(21):2013–23. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
