

Model selection may not be a mandatory step for phylogeny reconstruction
- PMID: 30804347
- PMCID: PMC6389923
- DOI: 10.1038/s41467-019-08822-w
Model selection may not be a mandatory step for phylogeny reconstruction
Abstract
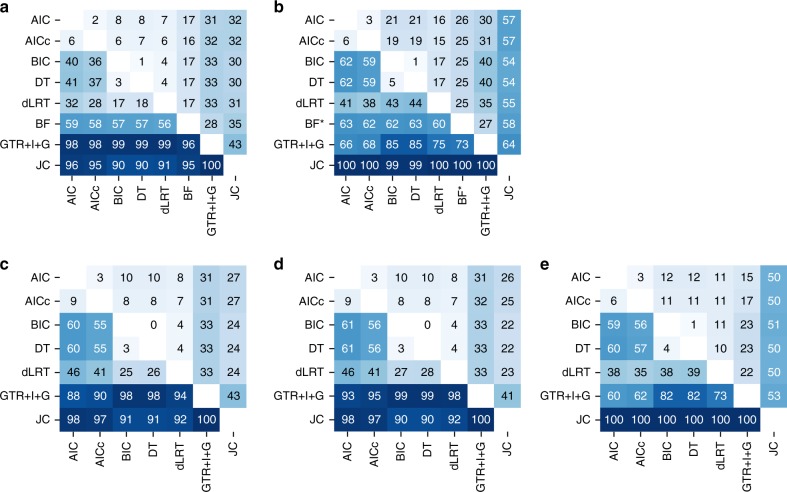
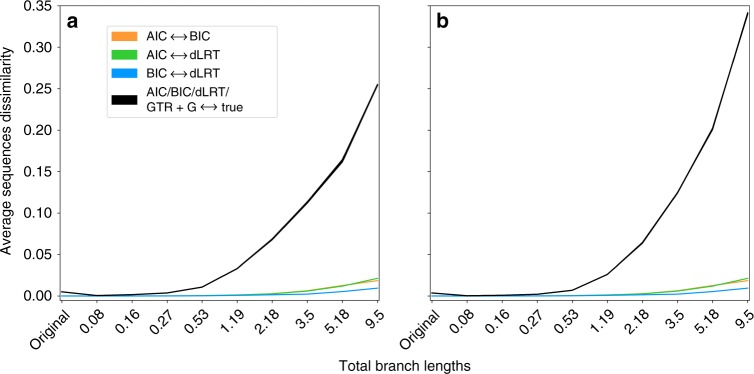
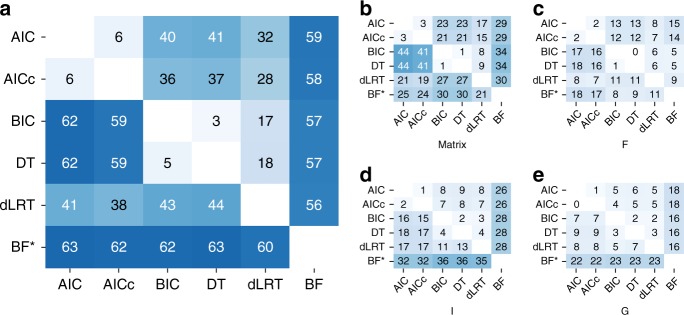
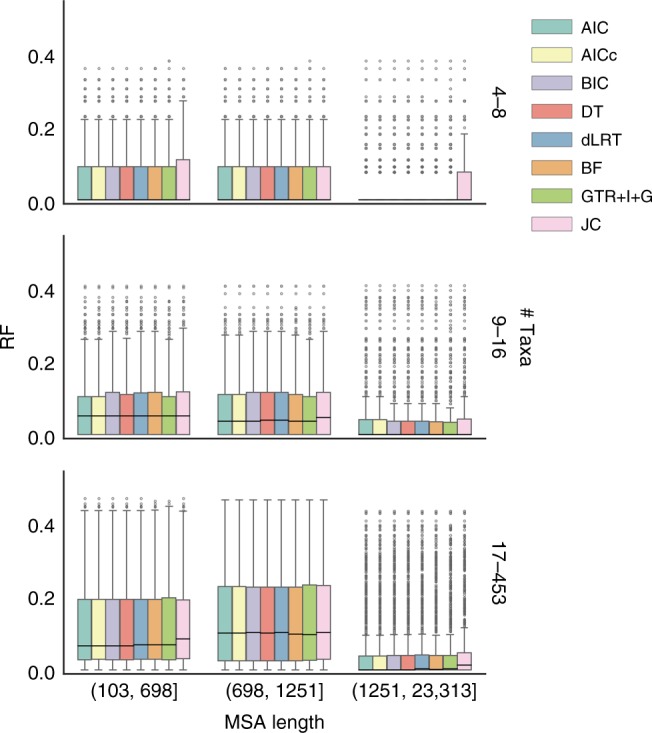
Determining the most suitable model for phylogeny reconstruction constitutes a fundamental step in numerous evolutionary studies. Over the years, various criteria for model selection have been proposed, leading to debate over which criterion is preferable. However, the necessity of this procedure has not been questioned to date. Here, we demonstrate that although incongruency regarding the selected model is frequent over empirical and simulated data, all criteria lead to very similar inferences. When topologies and ancestral sequence reconstruction are the desired output, choosing one criterion over another is not crucial. Moreover, skipping model selection and using instead the most parameter-rich model, GTR+I+G, leads to similar inferences, thus rendering this time-consuming step nonessential, at least under current strategies of model selection.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Jukes, T. H. & Cantor, C. R. in Mammalian Protein Metabolism. 21–132 (Academic Press, Cambridge, 1969).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
