

Isothermal Analysis of ThermoFluor Data can readily provide Quantitative Binding Affinities
- PMID: 30804351
- PMCID: PMC6389909
- DOI: 10.1038/s41598-018-37072-x
Isothermal Analysis of ThermoFluor Data can readily provide Quantitative Binding Affinities
Abstract
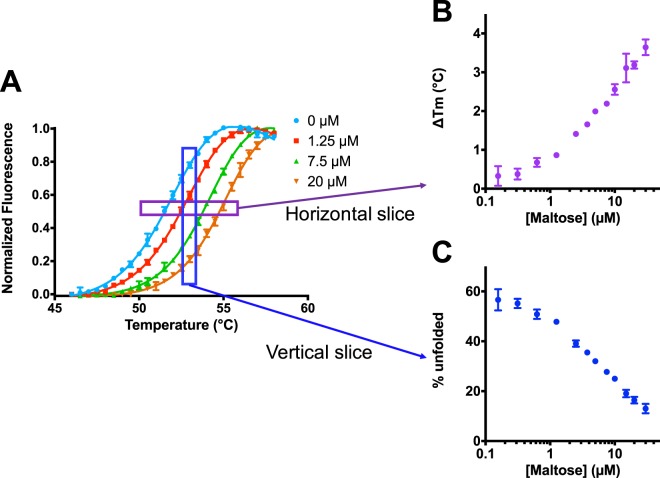
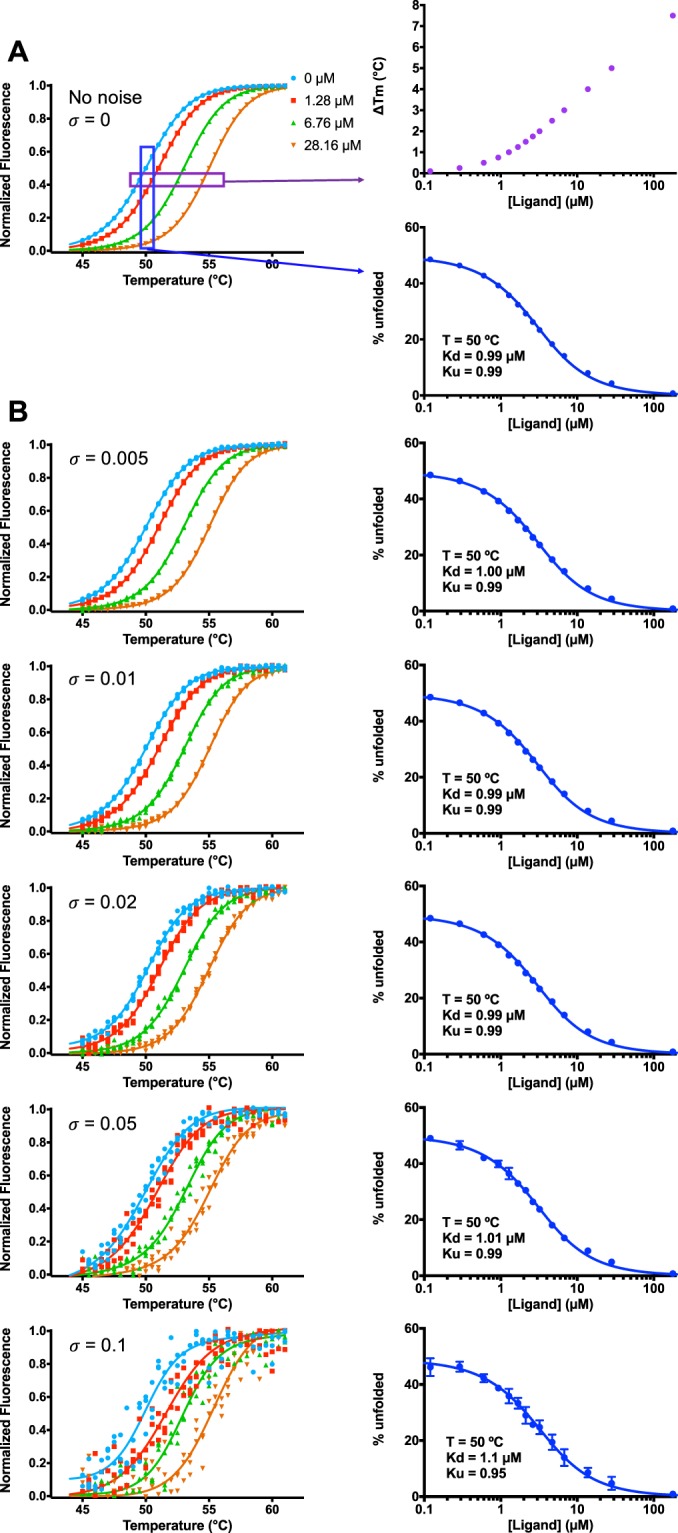
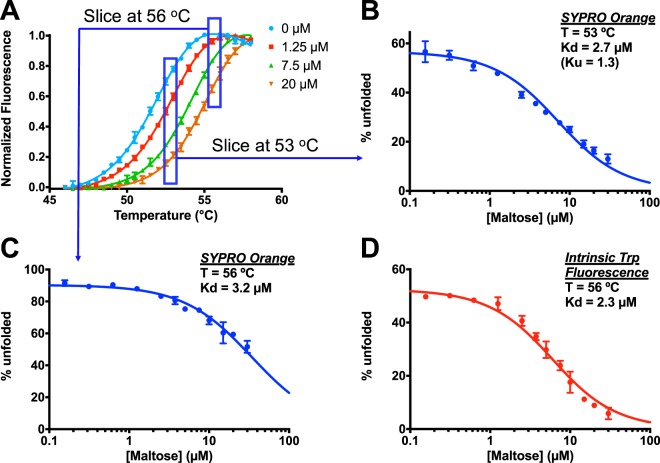
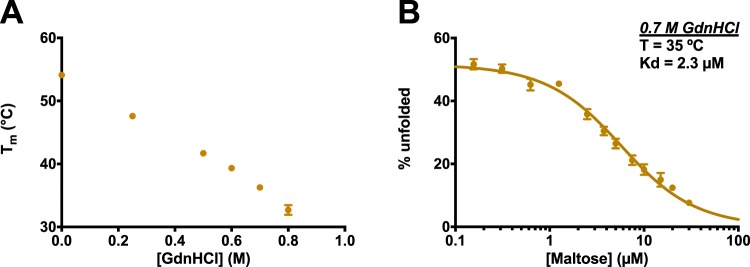
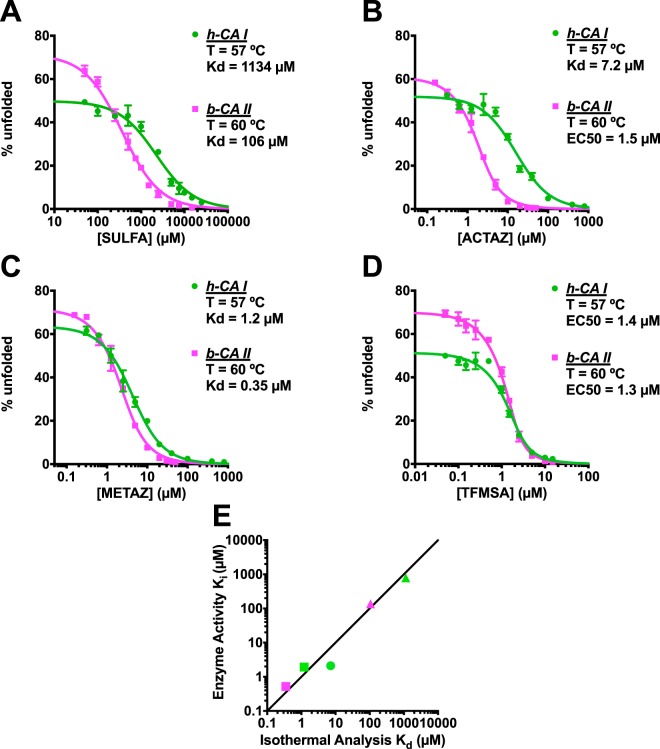
Differential scanning fluorimetry (DSF), also known as ThermoFluor or Thermal Shift Assay, has become a commonly-used approach for detecting protein-ligand interactions, particularly in the context of fragment screening. Upon binding to a folded protein, most ligands stabilize the protein; thus, observing an increase in the temperature at which the protein unfolds as a function of ligand concentration can serve as evidence of a direct interaction. While experimental protocols for this assay are well-developed, it is not straightforward to extract binding constants from the resulting data. Because of this, DSF is often used to probe for an interaction, but not to quantify the corresponding binding constant (Kd). Here, we propose a new approach for analyzing DSF data. Using unfolding curves at varying ligand concentrations, our "isothermal" approach collects from these the fraction of protein that is folded at a single temperature (chosen to be temperature near the unfolding transition). This greatly simplifies the subsequent analysis, because it circumvents the complicating temperature dependence of the binding constant; the resulting constant-temperature system can then be described as a pair of coupled equilibria (protein folding/unfolding and ligand binding/unbinding). The temperature at which the binding constants are determined can also be tuned, by adding chemical denaturants that shift the protein unfolding temperature. We demonstrate the application of this isothermal analysis using experimental data for maltose binding protein binding to maltose, and for two carbonic anhydrase isoforms binding to each of four inhibitors. To facilitate adoption of this new approach, we provide a free and easy-to-use Python program that analyzes thermal unfolding data and implements the isothermal approach described herein ( https://sourceforge.net/projects/dsf-fitting ).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Zhang R, Monsma F. Fluorescence-based thermal shift assays. Curr Opin Drug Discov Devel. 2010;13:389–402. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
