

Cellular TRIM33 restrains HIV-1 infection by targeting viral integrase for proteasomal degradation
- PMID: 30804369
- PMCID: PMC6389893
- DOI: 10.1038/s41467-019-08810-0
Cellular TRIM33 restrains HIV-1 infection by targeting viral integrase for proteasomal degradation
Abstract
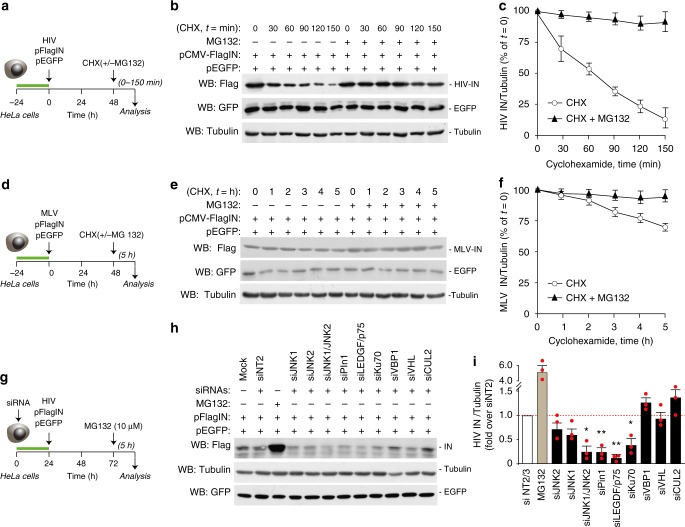
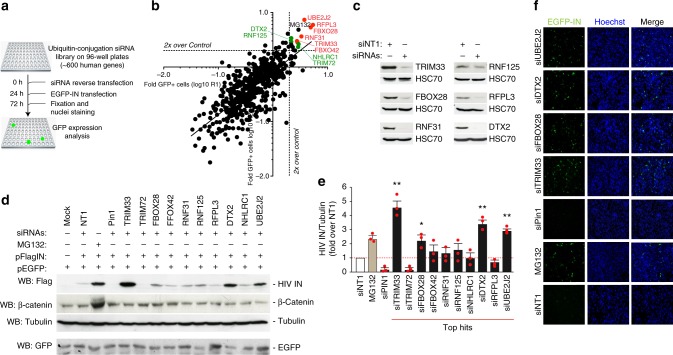
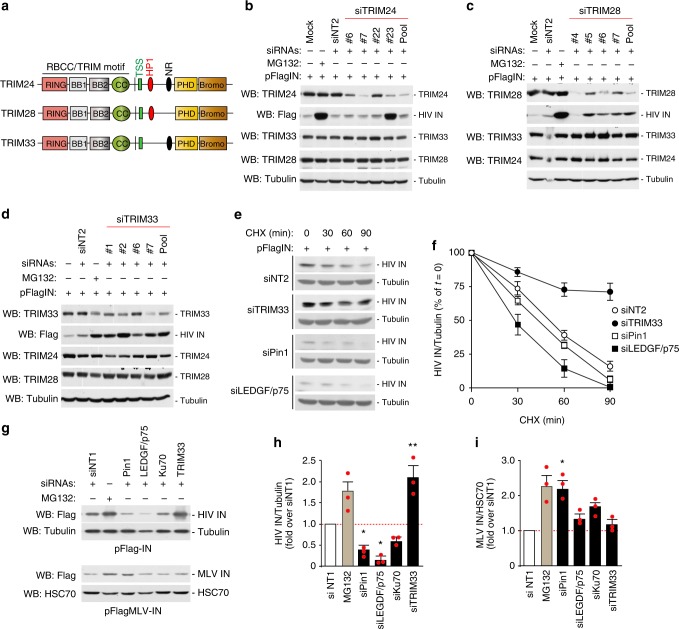
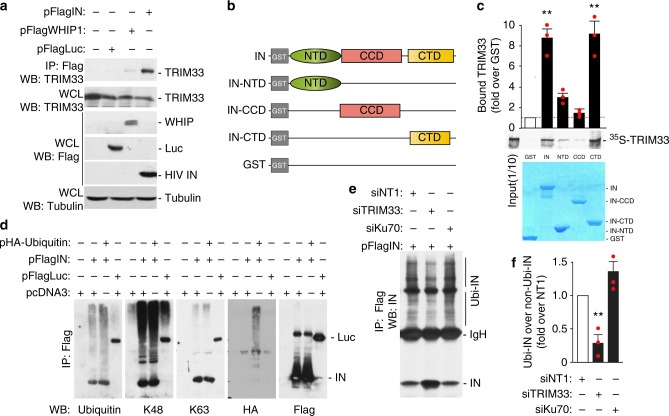
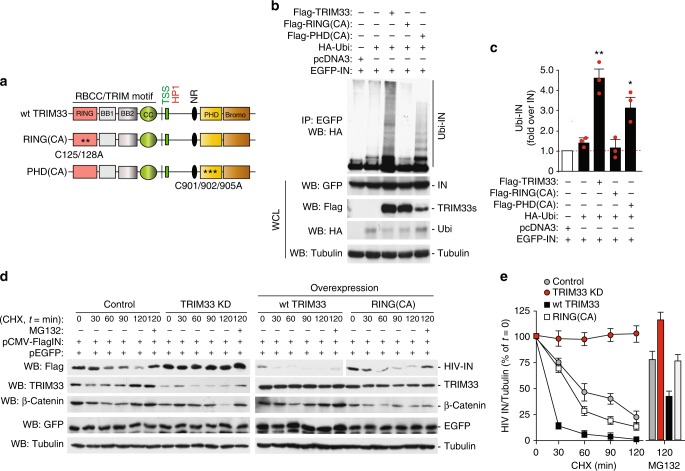
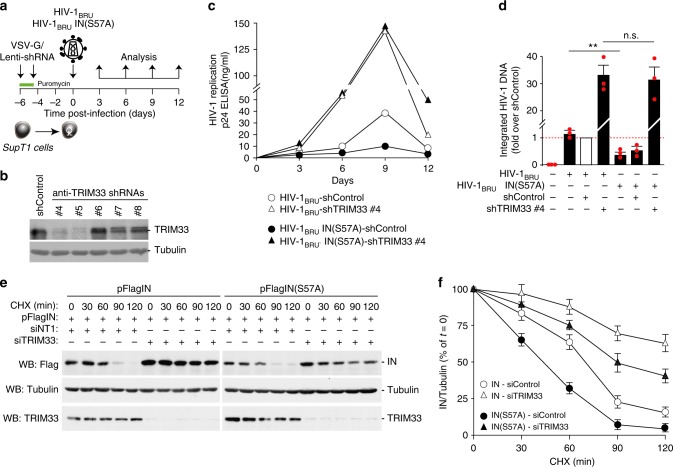
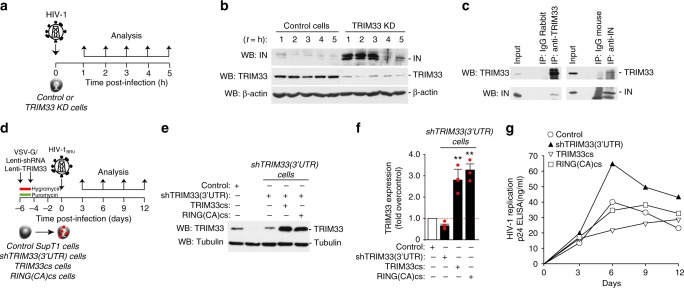
Productive HIV-1 replication requires viral integrase (IN), which catalyzes integration of the viral genome into the host cell DNA. IN, however, is short lived and is rapidly degraded by the host ubiquitin-proteasome system. To identify the cellular factors responsible for HIV-1 IN degradation, we performed a targeted RNAi screen using a library of siRNAs against all components of the ubiquitin-conjugation machinery using high-content microscopy. Here we report that the E3 RING ligase TRIM33 is a major determinant of HIV-1 IN stability. CD4-positive cells with TRIM33 knock down show increased HIV-1 replication and proviral DNA formation, while those overexpressing the factor display opposite effects. Knock down of TRIM33 reverts the phenotype of an HIV-1 molecular clone carrying substitution of IN serine 57 to alanine, a mutation known to impair viral DNA integration. Thus, TRIM33 acts as a cellular factor restricting HIV-1 infection by preventing provirus formation.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
