

Insights into the antigenic advancement of influenza A(H3N2) viruses, 2011-2018
- PMID: 30804469
- PMCID: PMC6389938
- DOI: 10.1038/s41598-019-39276-1
Insights into the antigenic advancement of influenza A(H3N2) viruses, 2011-2018
Abstract
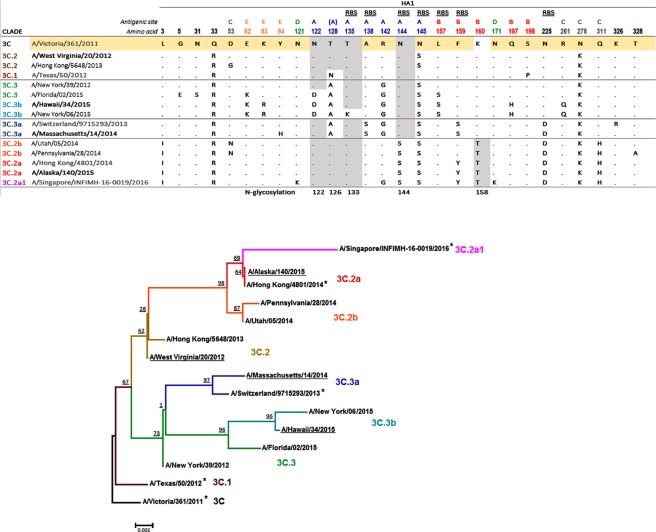
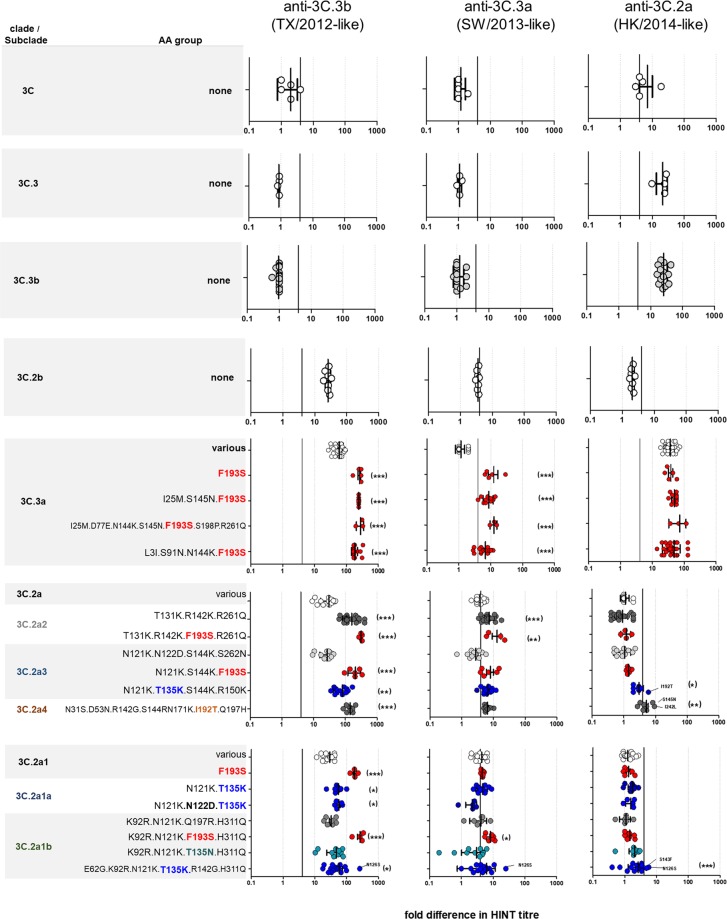
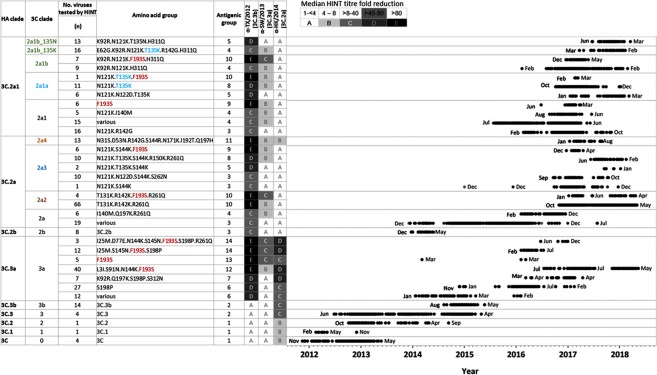
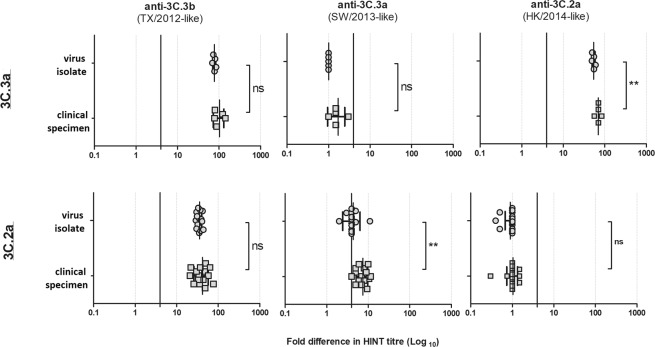
Influenza A(H3N2) viruses evade human immunity primarily by acquiring antigenic changes in the haemagglutinin (HA). HA receptor-binding features of contemporary A(H3N2) viruses hinder traditional antigenic characterization using haemagglutination inhibition and promote selection of HA mutants. Thus, alternative approaches are needed to reliably assess antigenic relatedness between circulating viruses and vaccines. We developed a high content imaging-based neutralization test (HINT) to reduce antigenic mischaracterization resulting from virus adaptation to cell culture. Ferret reference antisera were raised using clinical specimens containing viruses representing recent vaccine strains. Analysis of viruses circulating during 2011-2018 showed that gain of an N158-linked glycosylation in HA was a molecular determinant of antigenic distancing between A/Hong Kong/4801/2014-like (clade 3C.2a) and A/Texas/50/2012-like viruses (clade 3C.1), while multiple evolutionary HA F193S substitution were linked to antigenic distancing from A/Switzerland/97152963/2013-like (clade 3C.3a) and further antigenic distancing from A/Texas/50/2012-like viruses. Additionally, a few viruses carrying HA T135K and/or I192T showed reduced neutralization by A/Hong Kong/4801/2014-like antiserum. Notably, this technique elucidated the antigenic characteristics of clinical specimens, enabling direct characterization of viruses produced in vivo, and eliminating in vitro culture, which rapidly alters the genotype/phenotype. HINT is a valuable new antigenic analysis tool for vaccine strain selection.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Bedford, T. N. R. Seasonal influenza circulation patterns and projections for Feb 2018 to Feb 2019. bioRxiv (2018).
-
- WHO. Worldwide Influenza Centre- The Francis Crick Instititute. February 2017 Interim Report. Available from, https://www.crick.ac.uk/sites/default/files/2018-07/crick_sh2017_vcm_rep... (2017).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
