

Crosstalk Between Oxidative Stress and Endoplasmic Reticulum (ER) Stress in Endothelial Dysfunction and Aberrant Angiogenesis Associated With Diabetes: A Focus on the Protective Roles of Heme Oxygenase (HO)-1
- PMID: 30804804
- PMCID: PMC6378556
- DOI: 10.3389/fphys.2019.00070
Crosstalk Between Oxidative Stress and Endoplasmic Reticulum (ER) Stress in Endothelial Dysfunction and Aberrant Angiogenesis Associated With Diabetes: A Focus on the Protective Roles of Heme Oxygenase (HO)-1
Abstract
Type-2 diabetes prevalence is continuing to rise worldwide due to physical inactivity and obesity epidemic. Diabetes and fluctuations of blood sugar are related to multiple micro- and macrovascular complications, that are attributed to oxidative stress, endoplasmic reticulum (ER) activation and inflammatory processes, which lead to endothelial dysfunction characterized, among other features, by reduced availability of nitric oxide (NO) and aberrant angiogenic capacity. Several enzymatic anti-oxidant and anti-inflammatory agents have been found to play protective roles against oxidative stress and its downstream signaling pathways. Of particular interest, heme oxygenase (HO) isoforms, specifically HO-1, have attracted much attention as major cytoprotective players in conditions associated with inflammation and oxidative stress. HO operates as a key rate-limiting enzyme in the process of degradation of the iron-containing molecule, heme, yielding the following byproducts: carbon monoxide (CO), iron, and biliverdin. Because HO-1 induction was linked to pro-oxidant states, it has been regarded as a marker of oxidative stress; however, accumulating evidence has established multiple cytoprotective roles of the enzyme in metabolic and cardiovascular disorders. The cytoprotective effects of HO-1 depend on several cellular mechanisms including the generation of bilirubin, an anti-oxidant molecule, from the degradation of heme; the induction of ferritin, a strong chelator of free iron; and the release of CO, that displays multiple anti-inflammatory and anti-apoptotic actions. The current review article describes the major molecular mechanisms contributing to endothelial dysfunction and altered angiogenesis in diabetes with a special focus on the interplay between oxidative stress and ER stress response. The review summarizes the key cytoprotective roles of HO-1 against hyperglycemia-induced endothelial dysfunction and aberrant angiogenesis and discusses the major underlying cellular mechanisms associated with its protective effects.
Keywords: ER stress; angiogenesis; diabetes; endothelial dysfunction; heme oxygenase-1 (Ho-1); hyperglycemia; oxidative stress.
Figures

 means inhibition.
means inhibition. means inhibition.
means inhibition. means inhibition.
means inhibition.


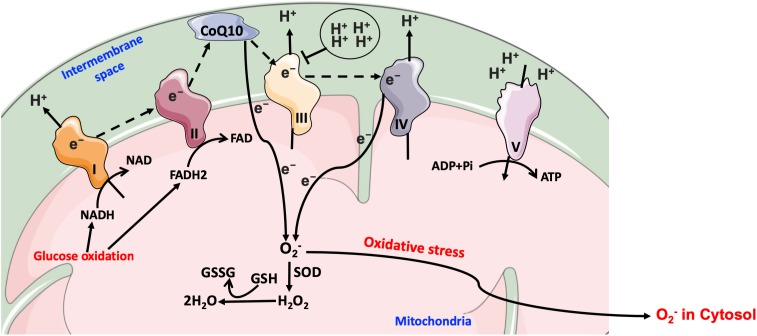
 means inhibition.
means inhibition.Similar articles
-
Heme oxygenase (HO)-1 induction prevents Endoplasmic Reticulum stress-mediated endothelial cell death and impaired angiogenic capacity.Biochem Pharmacol. 2017 Mar 1;127:46-59. doi: 10.1016/j.bcp.2016.12.009. Epub 2016 Dec 21. Biochem Pharmacol. 2017. PMID: 28012960
-
Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury.Am J Respir Cell Mol Biol. 1996 Jul;15(1):9-19. doi: 10.1165/ajrcmb.15.1.8679227. Am J Respir Cell Mol Biol. 1996. PMID: 8679227 Review.
-
Heme Degradation by Heme Oxygenase Protects Mitochondria but Induces ER Stress via Formed Bilirubin.Biomolecules. 2015 Apr 30;5(2):679-701. doi: 10.3390/biom5020679. Biomolecules. 2015. PMID: 25942605 Free PMC article.
-
Heme, Heme Oxygenase, and Endoplasmic Reticulum Stress-A New Insight into the Pathophysiology of Vascular Diseases.Int J Mol Sci. 2019 Jul 26;20(15):3675. doi: 10.3390/ijms20153675. Int J Mol Sci. 2019. PMID: 31357546 Free PMC article. Review.
-
Heme Oxygenase-1: An Anti-Inflammatory Effector in Cardiovascular, Lung, and Related Metabolic Disorders.Antioxidants (Basel). 2022 Mar 15;11(3):555. doi: 10.3390/antiox11030555. Antioxidants (Basel). 2022. PMID: 35326205 Free PMC article. Review.
Cited by
-
Porcine placenta extract improves high-glucose-induced angiogenesis impairment.BMC Complement Med Ther. 2021 Feb 18;21(1):66. doi: 10.1186/s12906-021-03243-z. BMC Complement Med Ther. 2021. PMID: 33602182 Free PMC article.
-
Endoplasmic Reticulum (ER) Stress-Generated Extracellular Vesicles (Microparticles) Self-Perpetuate ER Stress and Mediate Endothelial Cell Dysfunction Independently of Cell Survival.Front Cardiovasc Med. 2020 Dec 10;7:584791. doi: 10.3389/fcvm.2020.584791. eCollection 2020. Front Cardiovasc Med. 2020. PMID: 33363219 Free PMC article.
-
High Glucose-Induced Cardiomyocyte Damage Involves Interplay between Endothelin ET-1/ETA/ETB Receptor and mTOR Pathway.Int J Mol Sci. 2022 Nov 10;23(22):13816. doi: 10.3390/ijms232213816. Int J Mol Sci. 2022. PMID: 36430296 Free PMC article.
-
Construction of heparin-based hydrogel incorporated with Cu5.4O ultrasmall nanozymes for wound healing and inflammation inhibition.Bioact Mater. 2021 Mar 9;6(10):3109-3124. doi: 10.1016/j.bioactmat.2021.02.006. eCollection 2021 Oct. Bioact Mater. 2021. PMID: 33778192 Free PMC article.
-
Attenuation of PI3K/AKT signaling pathway by Ocimum gratissimum leaf flavonoid-rich extracts in streptozotocin-induced diabetic male rats.Biochem Biophys Rep. 2024 May 16;38:101735. doi: 10.1016/j.bbrep.2024.101735. eCollection 2024 Jul. Biochem Biophys Rep. 2024. PMID: 38799115 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources