

Long-term balancing selection drives evolution of immunity genes in Capsella
- PMID: 30806624
- PMCID: PMC6426441
- DOI: 10.7554/eLife.43606
Long-term balancing selection drives evolution of immunity genes in Capsella
Abstract
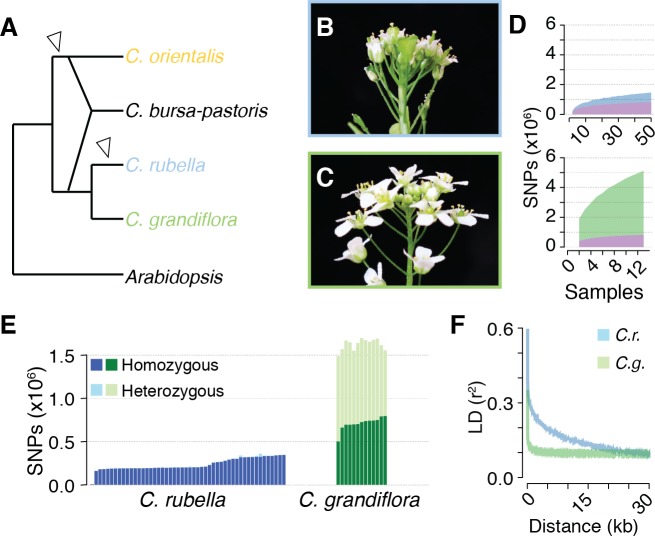
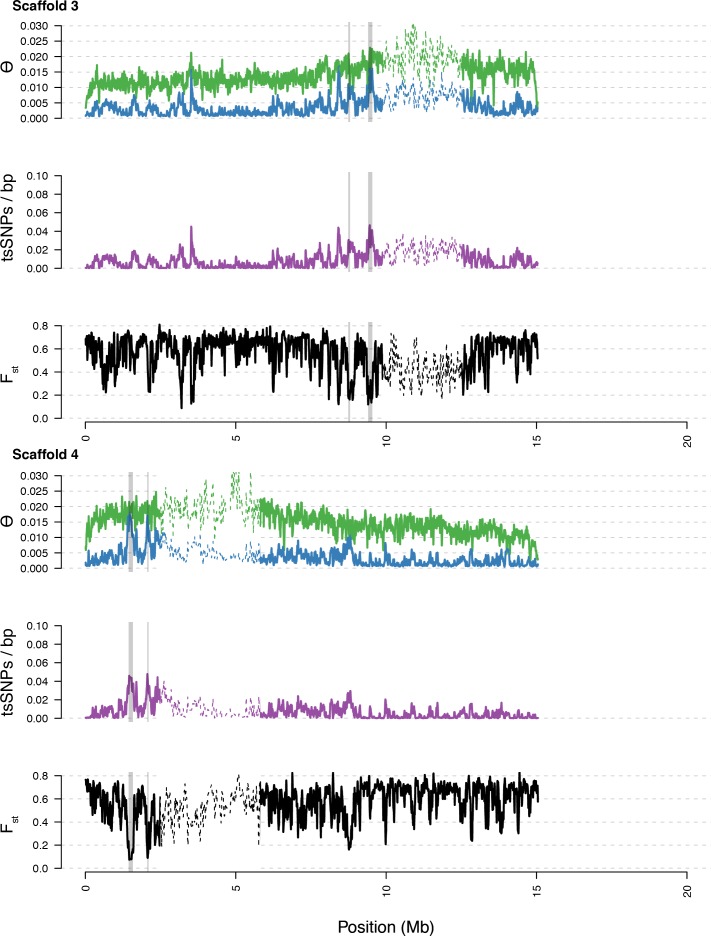
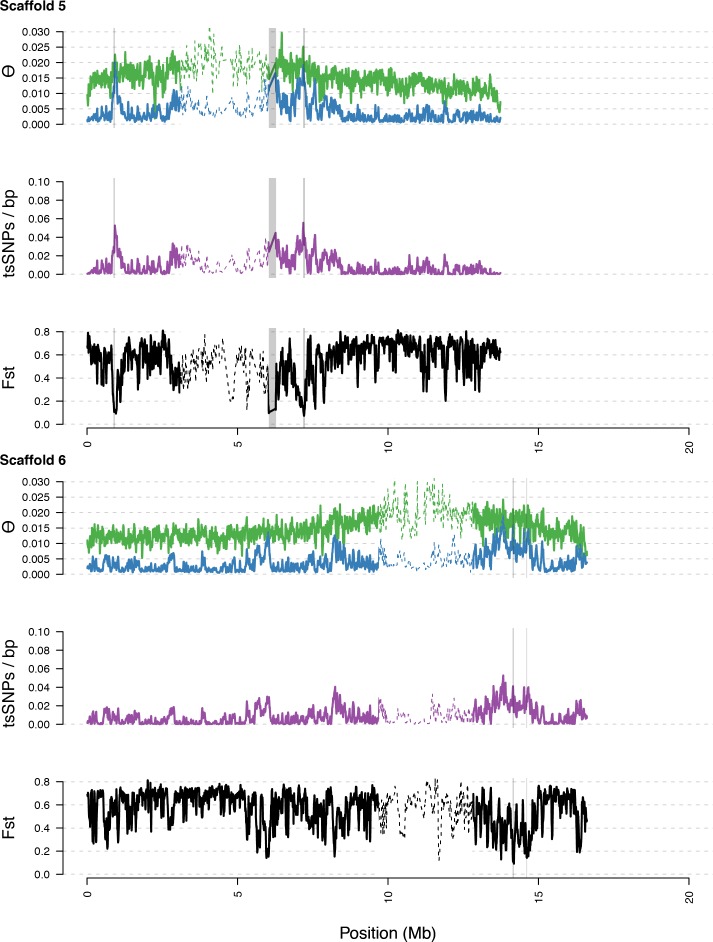
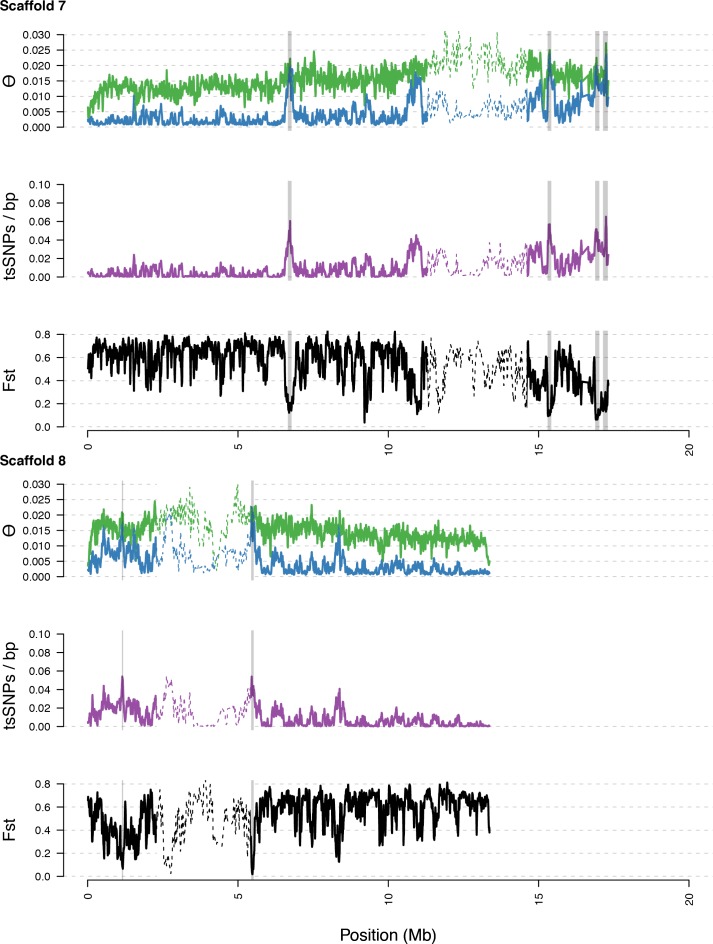
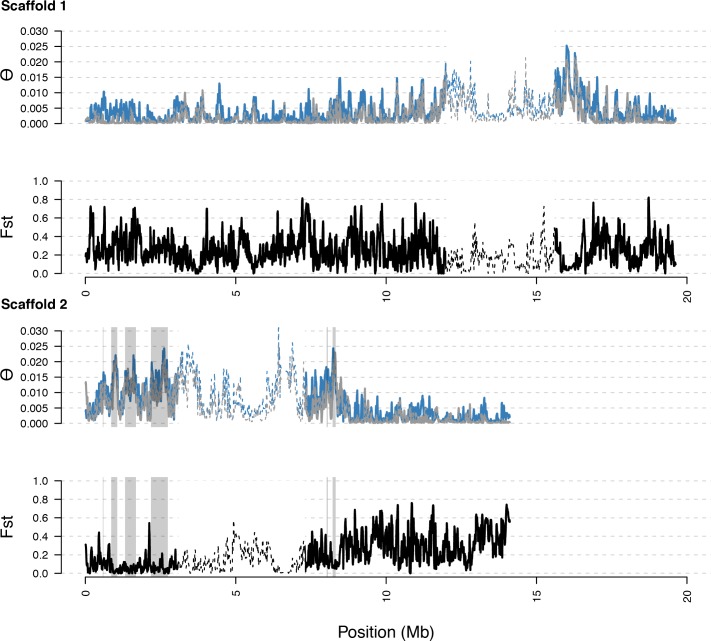
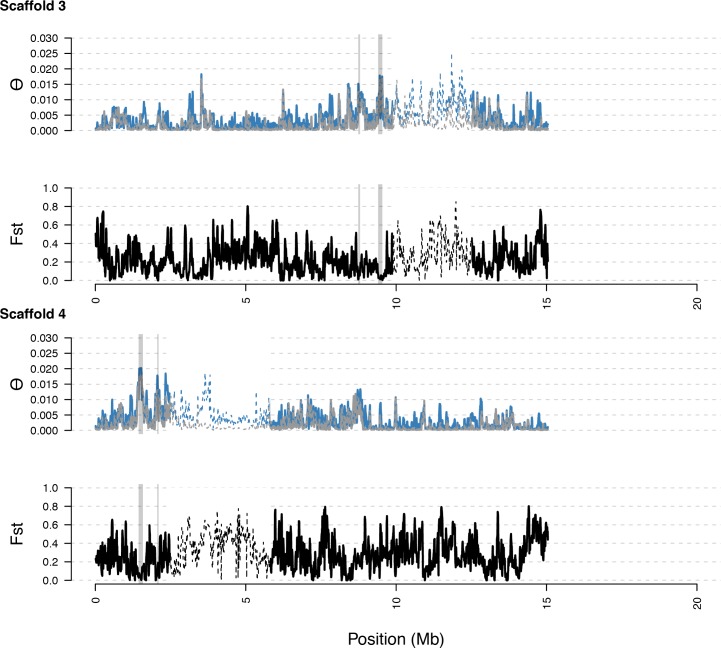
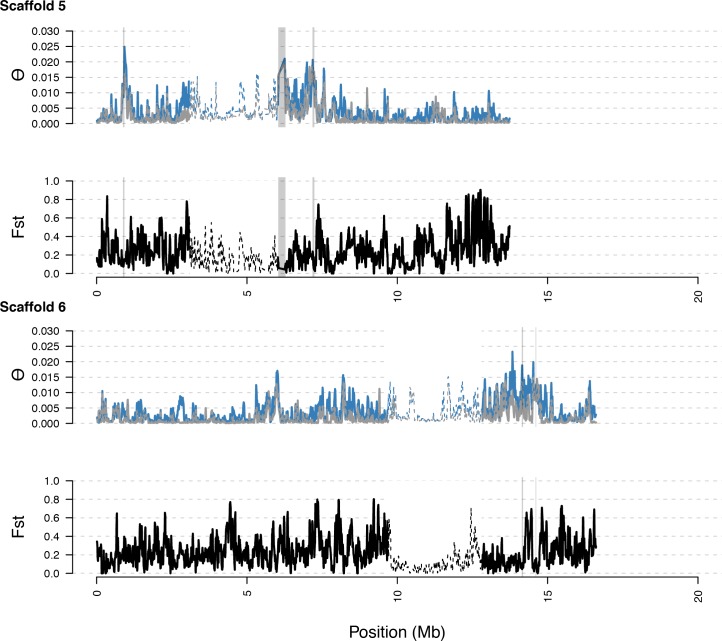
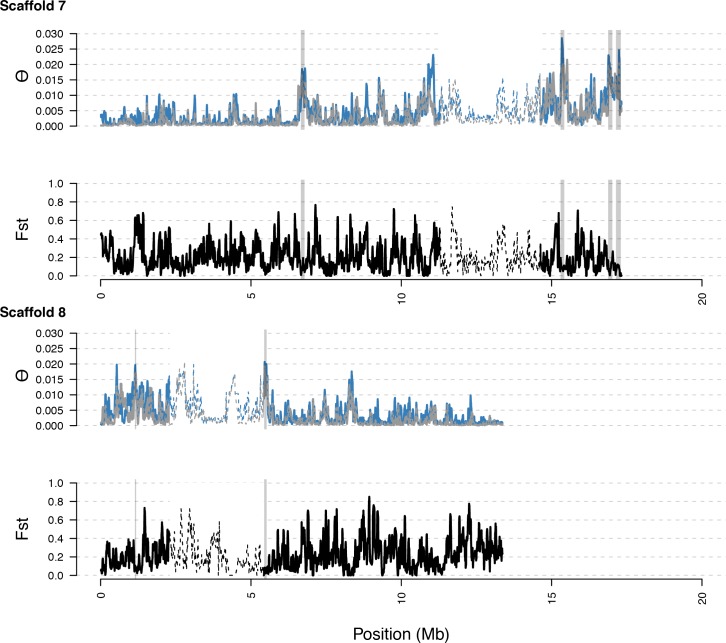
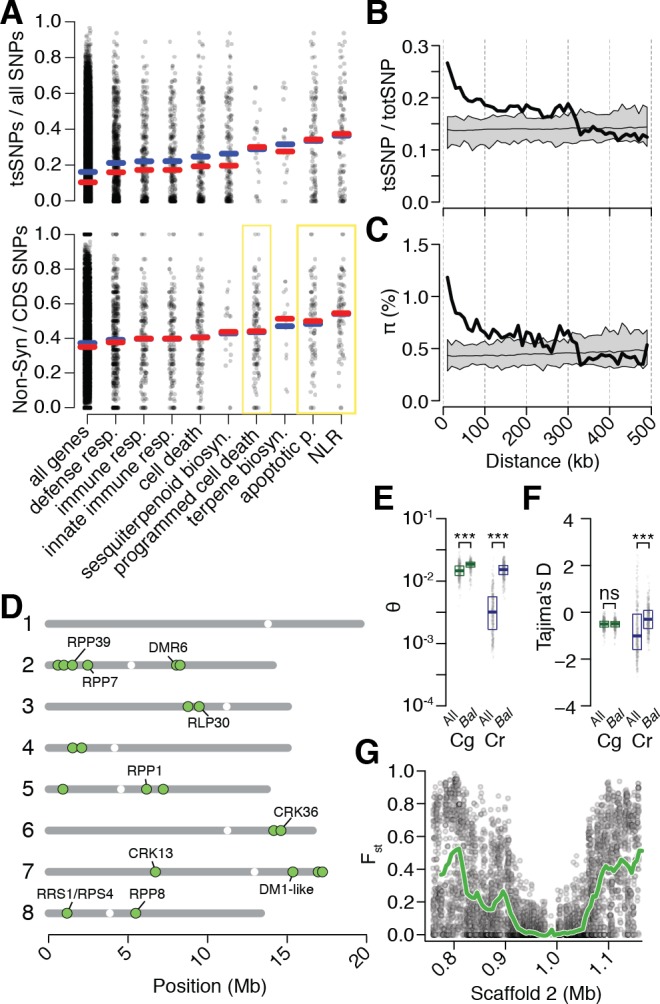
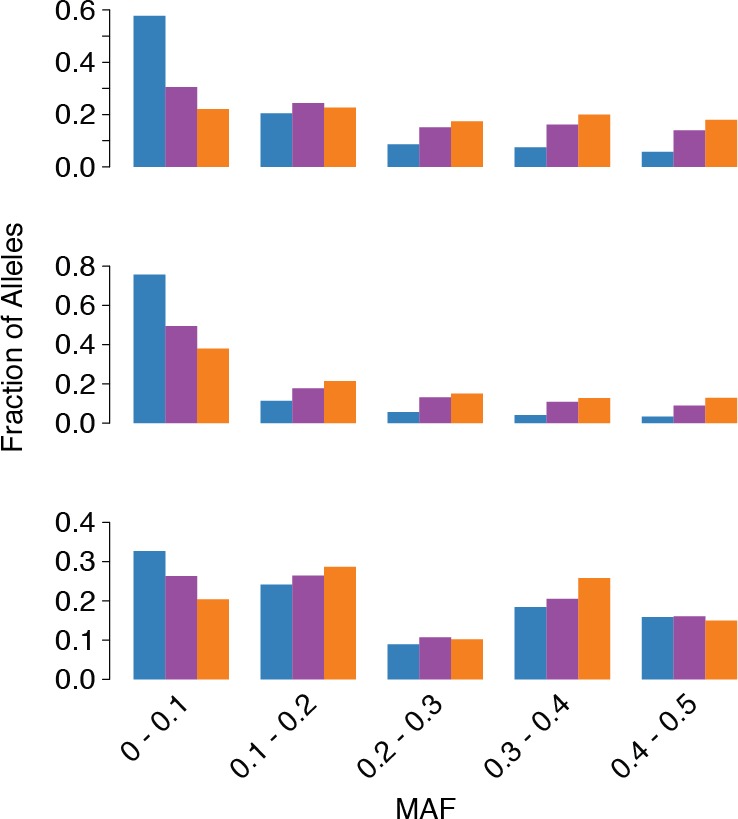
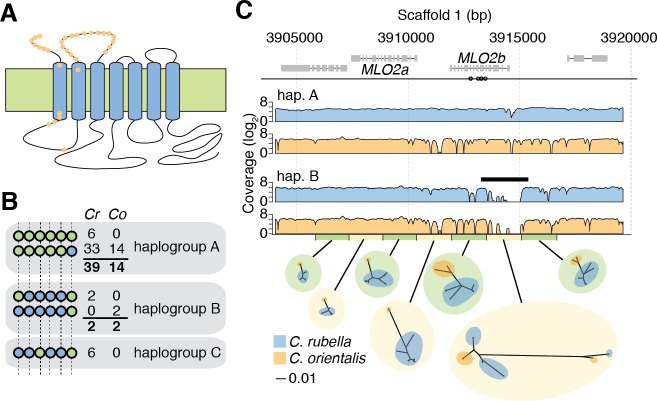
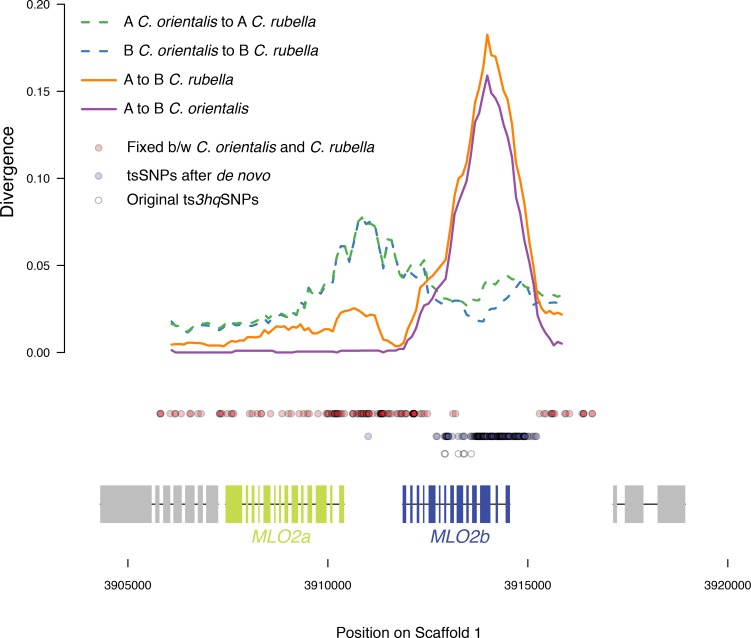
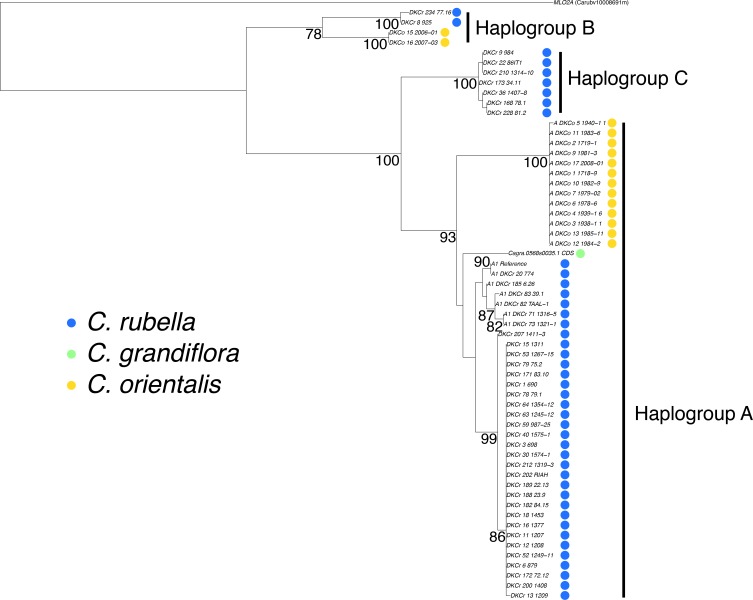
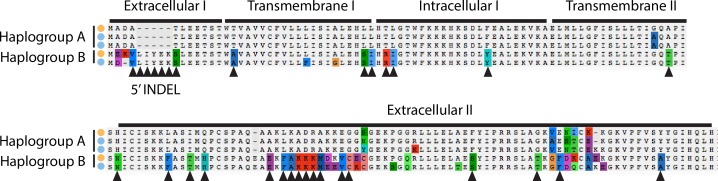
Genetic drift is expected to remove polymorphism from populations over long periods of time, with the rate of polymorphism loss being accelerated when species experience strong reductions in population size. Adaptive forces that maintain genetic variation in populations, or balancing selection, might counteract this process. To understand the extent to which natural selection can drive the retention of genetic diversity, we document genomic variability after two parallel species-wide bottlenecks in the genus Capsella. We find that ancestral variation preferentially persists at immunity related loci, and that the same collection of alleles has been maintained in different lineages that have been separated for several million years. By reconstructing the evolution of the disease-related locus MLO2b, we find that divergence between ancient haplotypes can be obscured by referenced based re-sequencing methods, and that trans-specific alleles can encode substantially diverged protein sequences. Our data point to long-term balancing selection as an important factor shaping the genetics of immune systems in plants and as the predominant driver of genomic variability after a population bottleneck.
Keywords: NB-LRR; balancing selection; bottleneck; disease resistance; evolutionary biology; genetics; genomics; immunity; selfing.
© 2019, Koenig et al.
Conflict of interest statement
DK, JH, RL, FB, TS, BN, SW No competing interests declared, DW Deputy editor, eLife
Figures
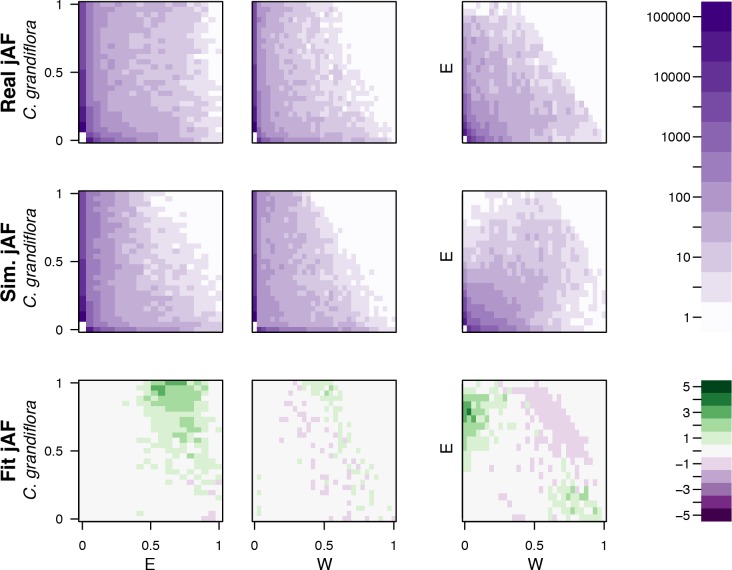
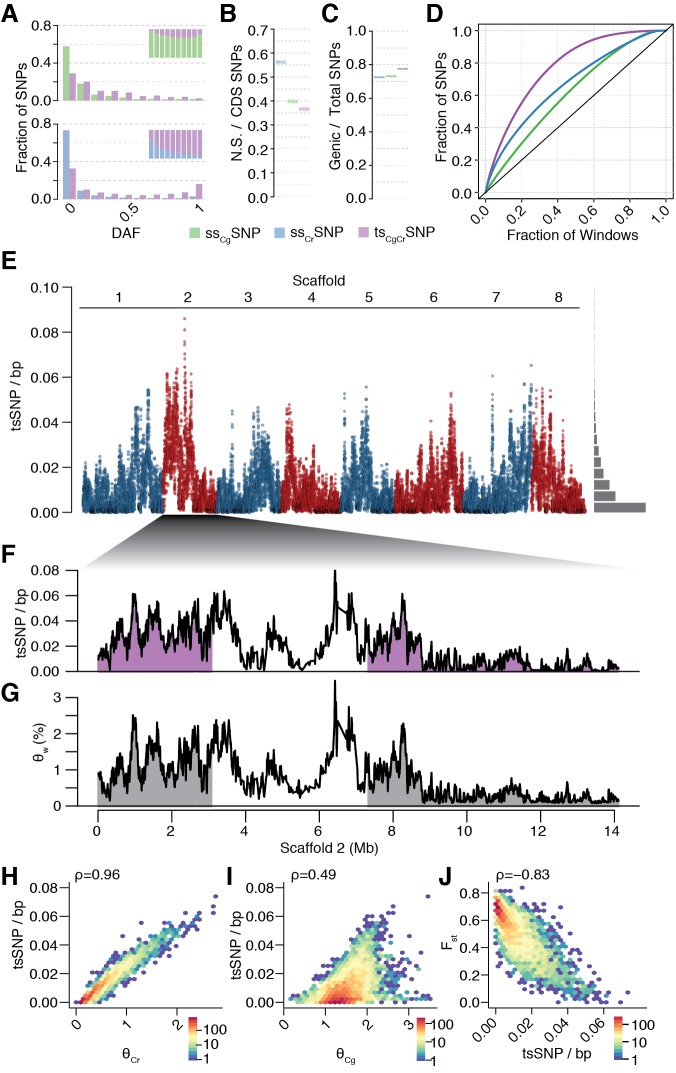
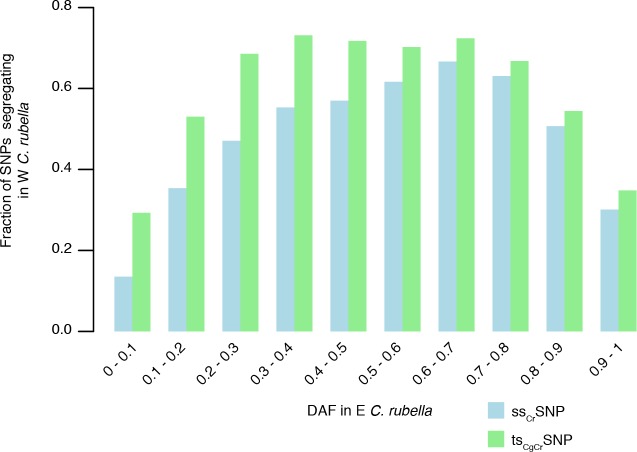
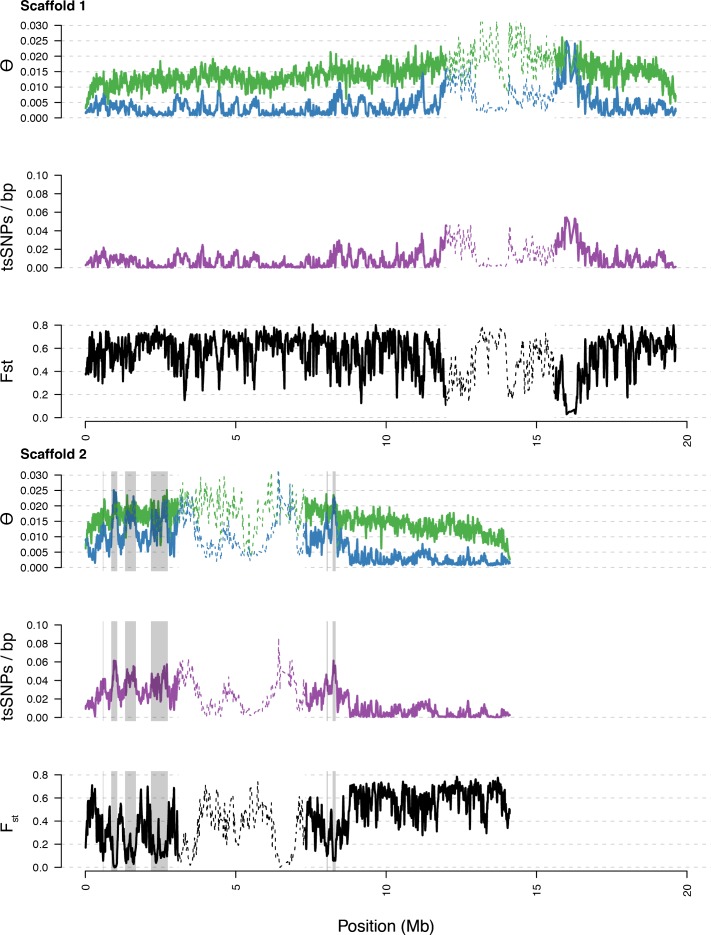
References
-
- Atwell S, Huang YS, Vilhjálmsson BJ, Willems G, Horton M, Li Y, Meng D, Platt A, Tarone AM, Hu TT, Jiang R, Muliyati NW, Zhang X, Amer MA, Baxter I, Brachi B, Chory J, Dean C, Debieu M, de Meaux J, Ecker JR, Faure N, Kniskern JM, Jones JD, Michael T, Nemri A, Roux F, Salt DE, Tang C, Todesco M, Traw MB, Weigel D, Marjoram P, Borevitz JO, Bergelson J, Nordborg M. Genome-wide association study of 107 phenotypes in arabidopsis thaliana inbred lines. Nature. 2010;465:627–631. doi: 10.1038/nature08800. - DOI - PMC - PubMed
