

Sunitinib promotes myogenic regeneration and mitigates disease progression in the mdx mouse model of Duchenne muscular dystrophy
- PMID: 30806670
- PMCID: PMC6586148
- DOI: 10.1093/hmg/ddz044
Sunitinib promotes myogenic regeneration and mitigates disease progression in the mdx mouse model of Duchenne muscular dystrophy
Abstract
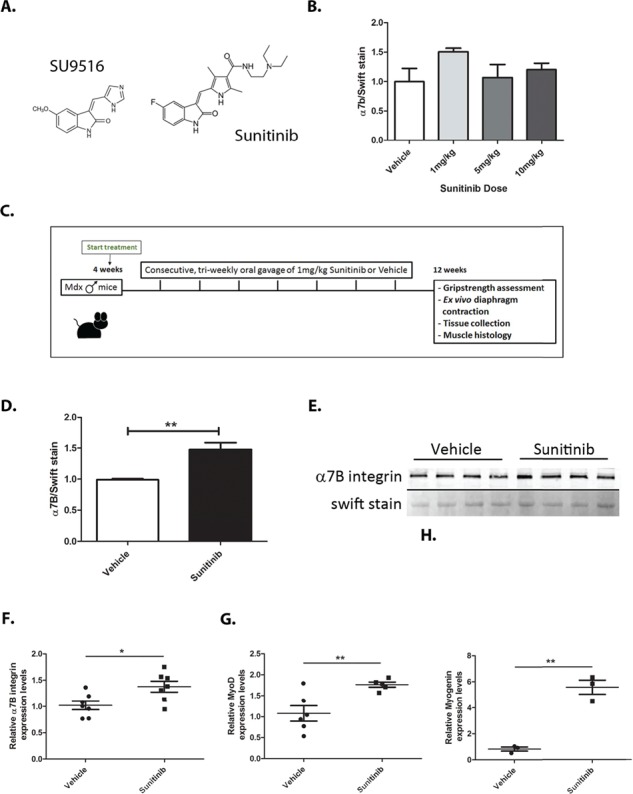
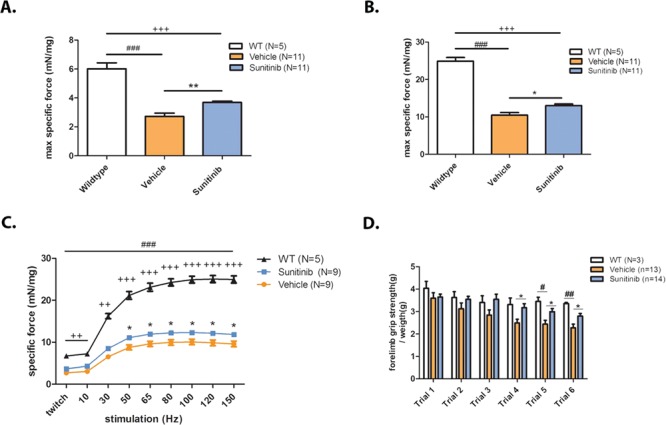
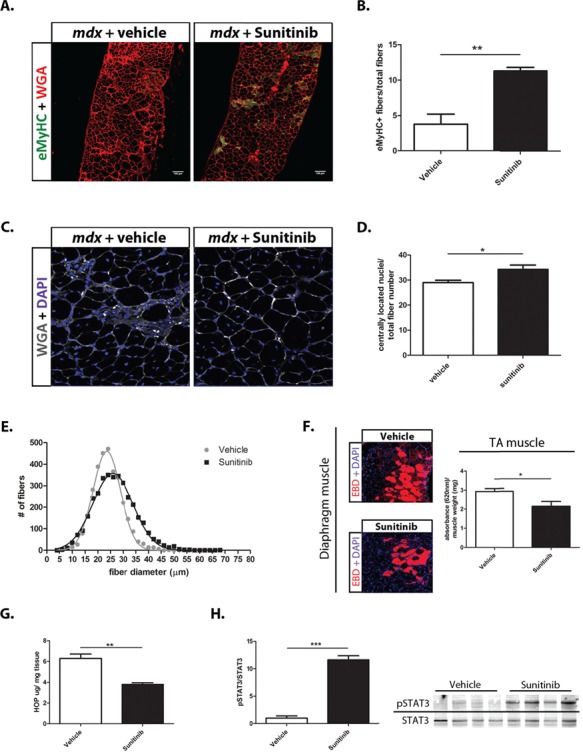
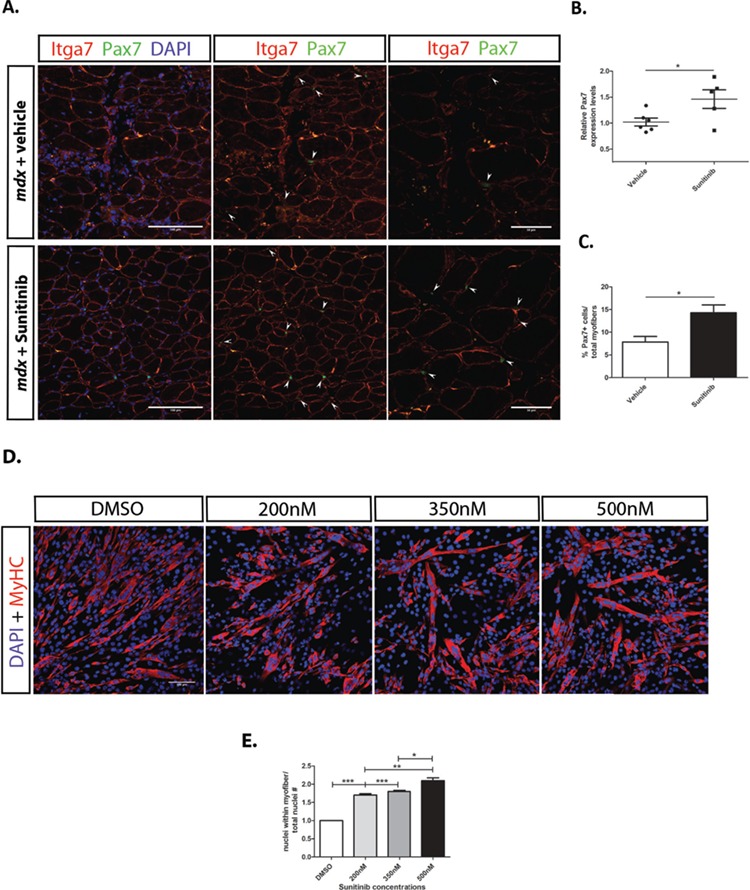
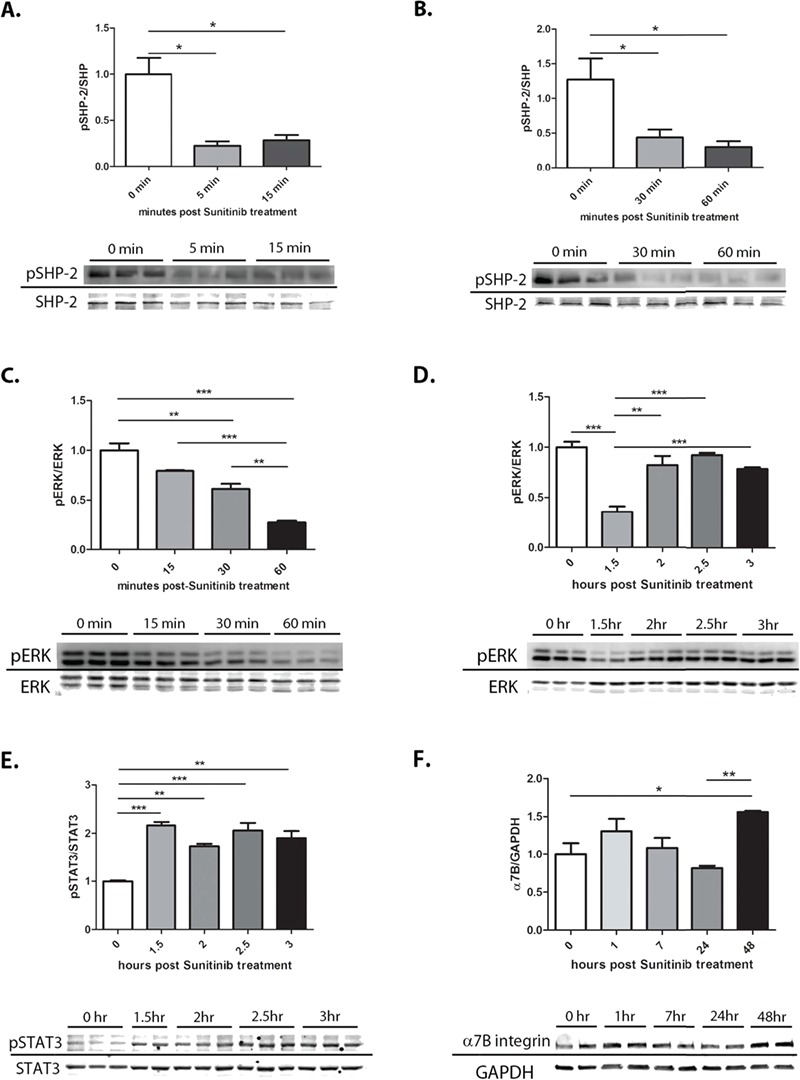
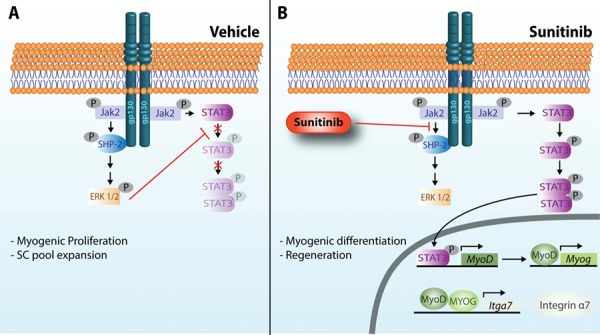
Duchenne muscular dystrophy (DMD) is a lethal, muscle degenerative disease causing premature death of affected children. DMD is characterized by mutations in the dystrophin gene that result in a loss of the dystrophin protein. Loss of dystrophin causes an associated reduction in proteins of the dystrophin glycoprotein complex, leading to contraction-induced sarcolemmal weakening, muscle tearing, fibrotic infiltration and rounds of degeneration and failed regeneration affecting satellite cell populations. The α7β1 integrin has been implicated in increasing myogenic capacity of satellite cells, therefore restoring muscle viability, increasing muscle force and preserving muscle function in dystrophic mouse models. In this study, we show that a Food and Drug Administration (FDA)-approved small molecule, Sunitinib, is a potent α7 integrin enhancer capable of promoting myogenic regeneration by stimulating satellite cell activation and increasing myofiber fusion. Sunitinib exerts its regenerative effects via transient inhibition of SHP-2 and subsequent activation of the STAT3 pathway. Treatment of mdx mice with Sunitinib demonstrated decreased membrane leakiness and damage owing to myofiber regeneration and enhanced support at the extracellular matrix. The decreased myofiber damage translated into a significant increase in muscle force production. This study identifies an already FDA-approved compound, Sunitinib, as a possible DMD therapeutic with the potential to treat other muscular dystrophies in which there is defective muscle repair.
© The Author(s) 2019. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
SU9516 Increases α7β1 Integrin and Ameliorates Disease Progression in the mdx Mouse Model of Duchenne Muscular Dystrophy.Mol Ther. 2017 Jun 7;25(6):1395-1407. doi: 10.1016/j.ymthe.2017.03.022. Epub 2017 Apr 5. Mol Ther. 2017. PMID: 28391962 Free PMC article.
-
Sunitinib inhibits STAT3 phosphorylation in cardiac muscle and prevents cardiomyopathy in the mdx mouse model of Duchenne muscular dystrophy.Hum Mol Genet. 2022 Jul 21;31(14):2358-2369. doi: 10.1093/hmg/ddac042. Hum Mol Genet. 2022. PMID: 35157045 Free PMC article.
-
Expression rate of myogenic regulatory factors and muscle growth factor after botulinum toxin A injection in the right masseter muscle of dystrophin deficient (mdx) mice.Adv Clin Exp Med. 2019 Jan;28(1):11-18. doi: 10.17219/acem/76263. Adv Clin Exp Med. 2019. PMID: 30085421
-
Satellite Cells in Muscular Dystrophy - Lost in Polarity.Trends Mol Med. 2016 Jun;22(6):479-496. doi: 10.1016/j.molmed.2016.04.002. Epub 2016 May 5. Trends Mol Med. 2016. PMID: 27161598 Free PMC article. Review.
-
Empowering Muscle Stem Cells for the Treatment of Duchenne Muscular Dystrophy.Cells Tissues Organs. 2022;211(6):641-654. doi: 10.1159/000514305. Epub 2021 Apr 28. Cells Tissues Organs. 2022. PMID: 33910206 Review.
Cited by
-
LncRNA-FKBP1C regulates muscle fiber type switching by affecting the stability of MYH1B.Cell Death Discov. 2021 Apr 9;7(1):73. doi: 10.1038/s41420-021-00463-7. Cell Death Discov. 2021. PMID: 33837177 Free PMC article.
-
PTPN1/2 inhibition promotes muscle stem cell differentiation in Duchenne muscular dystrophy.Life Sci Alliance. 2024 Oct 30;8(1):e202402831. doi: 10.26508/lsa.202402831. Print 2025 Jan. Life Sci Alliance. 2024. PMID: 39477543 Free PMC article.
-
Teaching an Old Molecule New Tricks: Drug Repositioning for Duchenne Muscular Dystrophy.Int J Mol Sci. 2019 Nov 30;20(23):6053. doi: 10.3390/ijms20236053. Int J Mol Sci. 2019. PMID: 31801292 Free PMC article. Review.
-
Multi-Modal Analysis of Satellite Cells Reveals Early Impairments at Pre-Contractile Stages of Myogenesis in Duchenne Muscular Dystrophy.Cells. 2025 Jun 13;14(12):892. doi: 10.3390/cells14120892. Cells. 2025. PMID: 40558519 Free PMC article.
-
MiR-208b Regulates the Conversion of Skeletal Muscle Fiber Types by Inhibiting Mettl8 Expression.Front Genet. 2022 Feb 23;13:820464. doi: 10.3389/fgene.2022.820464. eCollection 2022. Front Genet. 2022. PMID: 35281804 Free PMC article.
References
-
- Yiu E.M. and Kornberg A.J. (2015) Duchenne muscular dystrophy. J. Paediatr. Child Health, 51, 759–764. - PubMed
-
- Hoffman E.P., Brown R.H. and Kunkel L.M. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell, 51, 919–928. - PubMed
-
- Bonilla E., Samitt C.E., Miranda A.F., Hays A.P., Salviati G., DiMauro S., Kunkel L.M., Hoffman E.P. and Rowland L.P. (1988) Duchenne muscular dystrophy: deficiency of dystrophin at the muscle cell surface. Cell, 54, 447–452. - PubMed
-
- Durbeej M. and Campbell K.P. (2002) Muscular dystrophies involving the dystrophin–glycoprotein complex: an overview of current mouse models. Curr. Opin. Genet. Dev., 12, 349–361. - PubMed
-
- Le Rumeur E., Winder S.J. and Hubert J.F. (2010) Dystrophin: more than just the sum of its parts. Biochim. Biophys. Acta Proteins Proteom., 1804, 1713–1722. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
