

NOD1: An Interface Between Innate Immunity and Insulin Resistance
- PMID: 30807635
- PMCID: PMC6477778
- DOI: 10.1210/en.2018-01061
NOD1: An Interface Between Innate Immunity and Insulin Resistance
Abstract
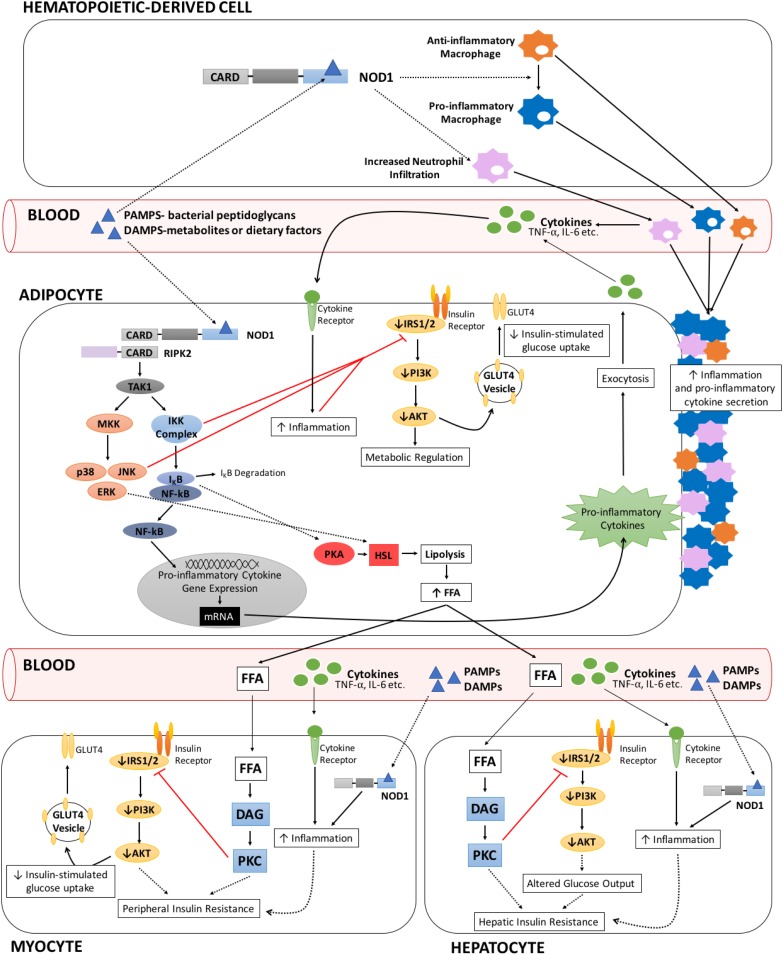
Insulin resistance is driven, in part, by activation of the innate immune system. We have discussed the evidence linking nucleotide-binding oligomerization domain (NOD)1, an intracellular pattern recognition receptor, to the onset and progression of obesity-induced insulin resistance. On a molecular level, crosstalk between downstream NOD1 effectors and the insulin receptor pathway inhibits insulin signaling, potentially through reduced insulin receptor substrate action. In vivo studies have demonstrated that NOD1 activation induces peripheral, hepatic, and whole-body insulin resistance. Also, NOD1-deficient models are protected from high-fat diet (HFD)-induced insulin resistance. Moreover, hematopoietic NOD1 deficiency prevented HFD-induced changes in proinflammatory macrophage polarization status, thus protecting against the development of metabolic inflammation and insulin resistance. Serum from HFD-fed mice activated NOD1 signaling ex vivo; however, the molecular identity of the activating factors remains unclear. Many have proposed that an HFD changes the gut permeability, resulting in increased translocation of bacterial fragments and increased circulating NOD1 ligands. In contrast, others have suggested that NOD1 ligands are endogenous and potentially lipid-derived metabolites produced during states of nutrient overload. Nevertheless, that NOD1 contributes to the development of insulin resistance, and that NOD1-based therapy might provide benefit, is an exciting advancement in metabolic research.
Copyright © 2019 Endocrine Society.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
