

Identification of transcription factor binding sites using ATAC-seq
- PMID: 30808370
- PMCID: PMC6391789
- DOI: 10.1186/s13059-019-1642-2
Identification of transcription factor binding sites using ATAC-seq
Abstract
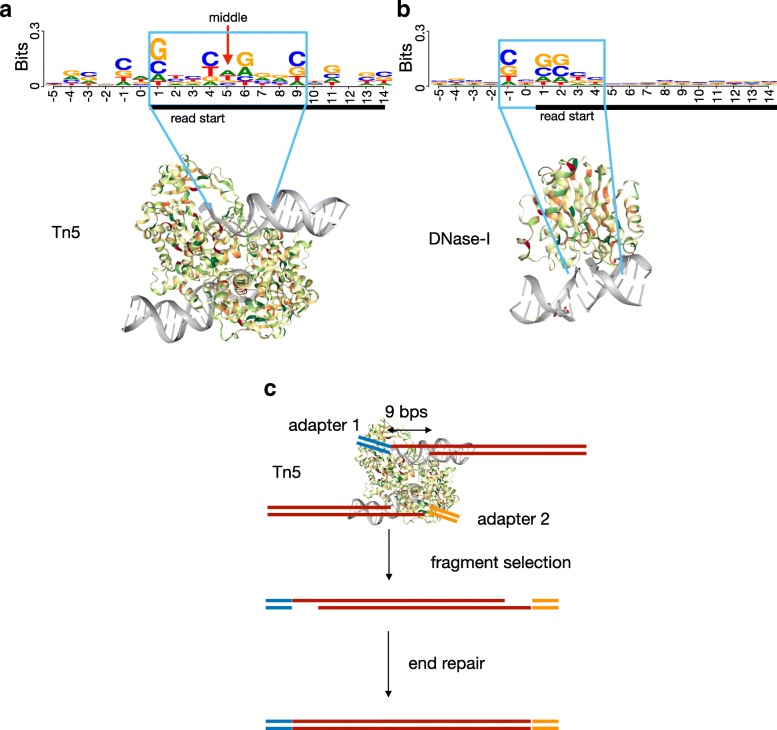
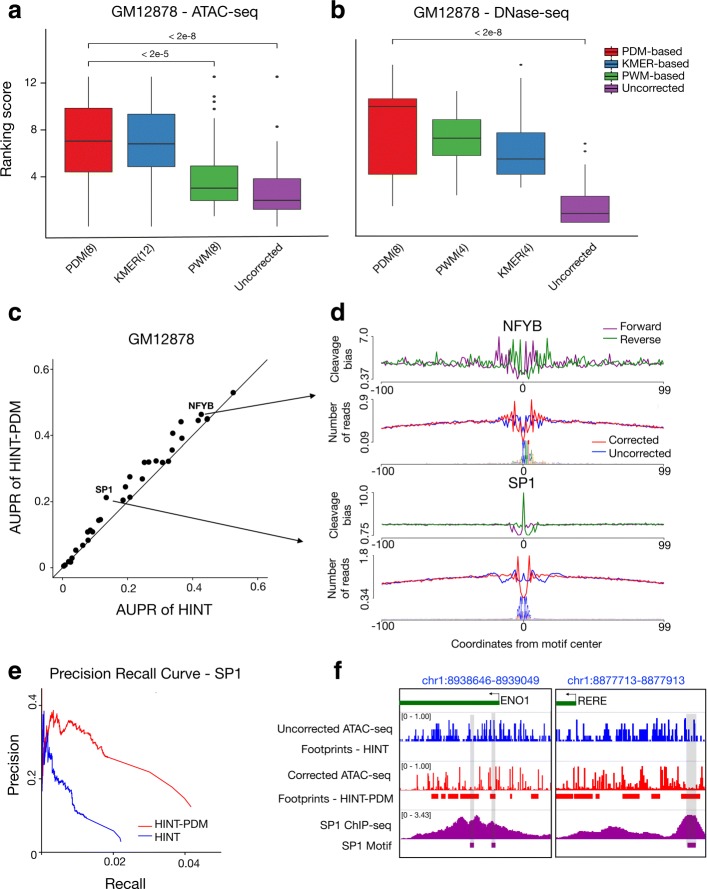
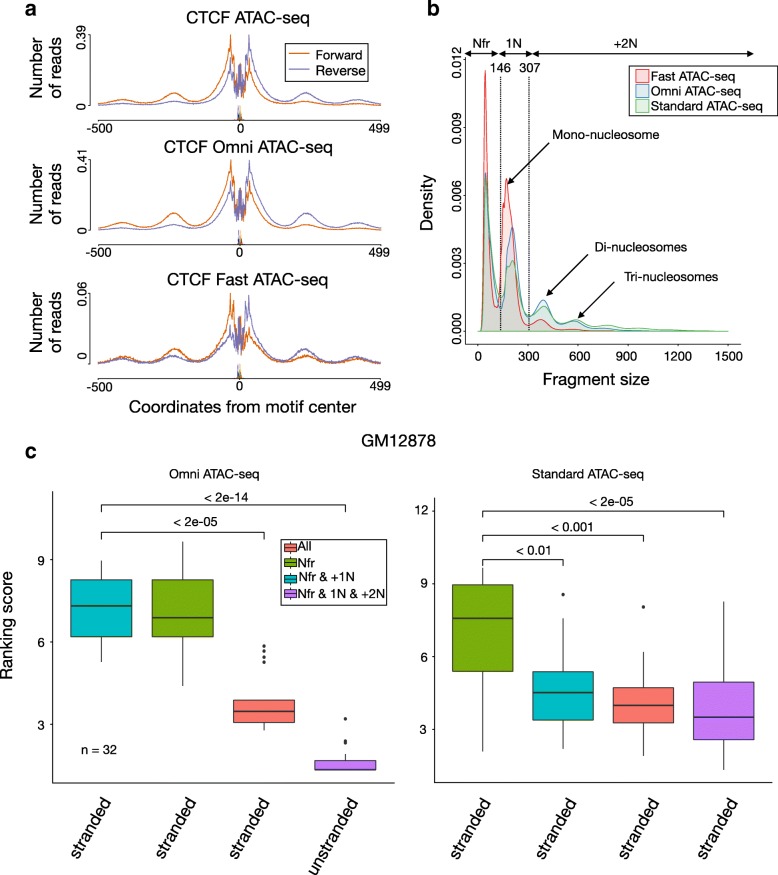
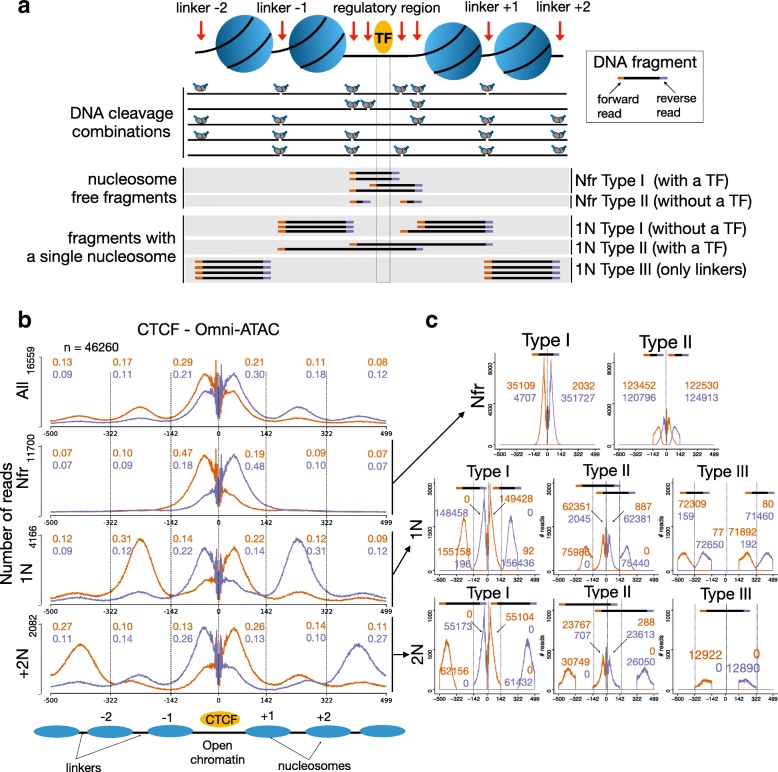
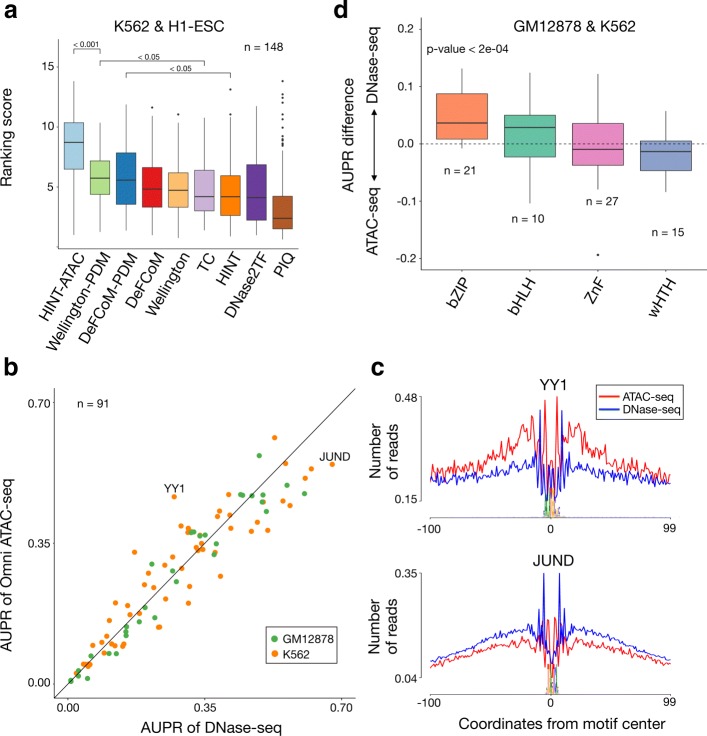
Transposase-Accessible Chromatin followed by sequencing (ATAC-seq) is a simple protocol for detection of open chromatin. Computational footprinting, the search for regions with depletion of cleavage events due to transcription factor binding, is poorly understood for ATAC-seq. We propose the first footprinting method considering ATAC-seq protocol artifacts. HINT-ATAC uses a position dependency model to learn the cleavage preferences of the transposase. We observe strand-specific cleavage patterns around transcription factor binding sites, which are determined by local nucleosome architecture. By incorporating all these biases, HINT-ATAC is able to significantly outperform competing methods in the prediction of transcription factor binding sites with footprints.
Keywords: ATAC-seq; Cleavage bias; Computational footprinting; Open chromatin.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Vierstra J, Stamatoyannopoulos JA. Nat Methods. 2016; 13(3):213–21. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
