

FGF23 and its role in X-linked hypophosphatemia-related morbidity
- PMID: 30808384
- PMCID: PMC6390548
- DOI: 10.1186/s13023-019-1014-8
FGF23 and its role in X-linked hypophosphatemia-related morbidity
Abstract
Background: X-linked hypophosphatemia (XLH) is an inherited disease of phosphate metabolism in which inactivating mutations of the Phosphate Regulating Endopeptidase Homolog, X-Linked (PHEX) gene lead to local and systemic effects including impaired growth, rickets, osteomalacia, bone abnormalities, bone pain, spontaneous dental abscesses, hearing difficulties, enthesopathy, osteoarthritis, and muscular dysfunction. Patients with XLH present with elevated levels of fibroblast growth factor 23 (FGF23), which is thought to mediate many of the aforementioned manifestations of the disease. Elevated FGF23 has also been observed in many other diseases of hypophosphatemia, and a range of animal models have been developed to study these diseases, yet the role of FGF23 in the pathophysiology of XLH is incompletely understood.
Methods: The role of FGF23 in the pathophysiology of XLH is here reviewed by describing what is known about phenotypes associated with various PHEX mutations, animal models of XLH, and non-nutritional diseases of hypophosphatemia, and by presenting molecular pathways that have been proposed to contribute to manifestations of XLH.
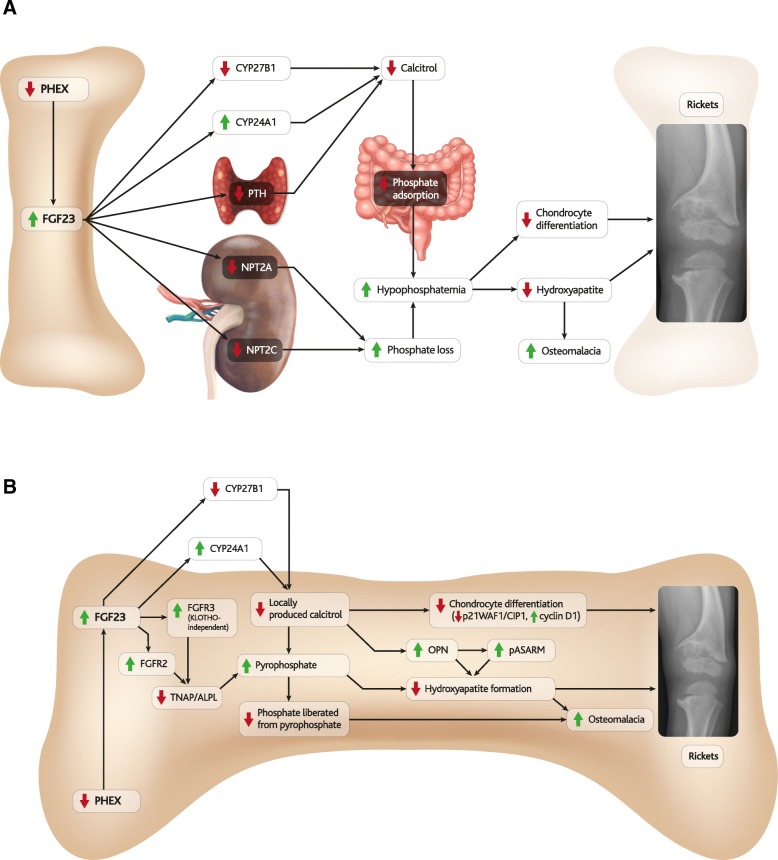
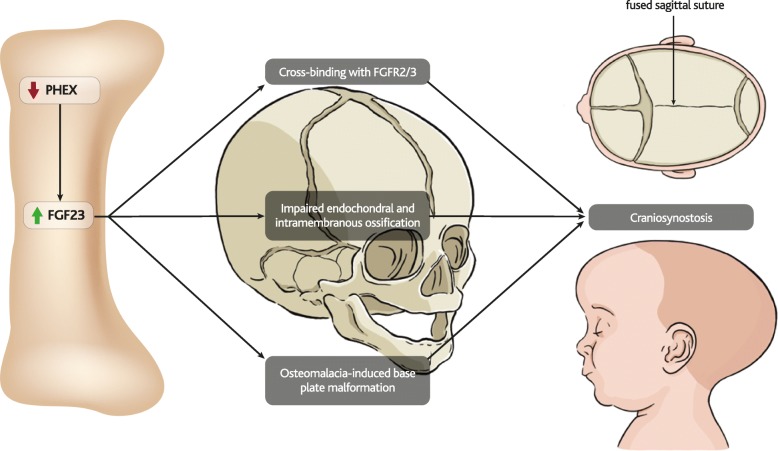
Results: The pathophysiology of XLH is complex, involving a range of molecular pathways that variously contribute to different manifestations of the disease. Hypophosphatemia due to elevated FGF23 is the most obvious contributor, however localised fluctuations in tissue non-specific alkaline phosphatase (TNAP), pyrophosphate, calcitriol and direct effects of FGF23 have been observed to be associated with certain manifestations.
Conclusions: By describing what is known about these pathways, this review highlights key areas for future research that would contribute to the understanding and clinical treatment of non-nutritional diseases of hypophosphatemia, particularly XLH.
Keywords: X-linked hypophosphatemia (XLH); bone dysplasia; dental abscess; ectopic calcification; fibroblast growth factor 23 (FGF23); hearing impairment; hypophosphatemia; muscle weakness; osteomalacia; phosphate regulating endopeptidase homolog, X-linked (PHEX); rickets; vitamin D deficiency.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
SBN, ZM, DH, ON, EL, GA, CDL, DS and OM have received honoraria and/or consultancy fees from Kyowa Kirin Services Ltd. DH and GA have received research support from Kyowa Kirin Services Ltd. SBN, DH and GA have received travel support from Kyowa Kirin Services Ltd. RJ is acting in a consultancy/advisory role for Kyowa Kirin Services Ltd.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures







References
-
- Beck-Nielsen SS, Brock-Jacobsen B, Gram J, Brixen K, Jensen TK. Incidence and prevalence of nutritional and hereditary rickets in southern Denmark. Eur J Endocrinol. 2009;160:491–497. - PubMed
-
- Reid IR, Hardy DC, Murphy WA, Teitelbaum SL, Bergfeld MA, Whyte MP. X-Linked Hypophosphatemia: A Clinical, Biochemical, and Histopathologic Assessment of Morbidity in Adults. Medicine (Baltimore) 1989;68:336. - PubMed
-
- Barros NM, Hoac B, Neves RL, Addison WN, Assis DM, Murshed M, et al. Proteolytic processing of osteopontin by PHEX and accumulation of osteopontin fragments in Hyp mouse bone, the murine model of X-linked hypophosphatemia. J Bone Min Res. 2013;28:688–699. - PubMed
-
- Liu S, Guo R, Simpson LG, Xiao Z-S, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003;278:37419–37426. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
