

Congenital myasthenic syndromes
- PMID: 30808424
- PMCID: PMC6390566
- DOI: 10.1186/s13023-019-1025-5
Congenital myasthenic syndromes
Abstract
Objectives: Congenital myasthenic syndromes (CMSs) are a genotypically and phenotypically heterogeneous group of neuromuscular disorders, which have in common an impaired neuromuscular transmission. Since the field of CMSs is steadily expanding, the present review aimed at summarizing and discussing current knowledge and recent advances concerning the etiology, clinical presentation, diagnosis, and treatment of CMSs.
Methods: Systematic literature review.
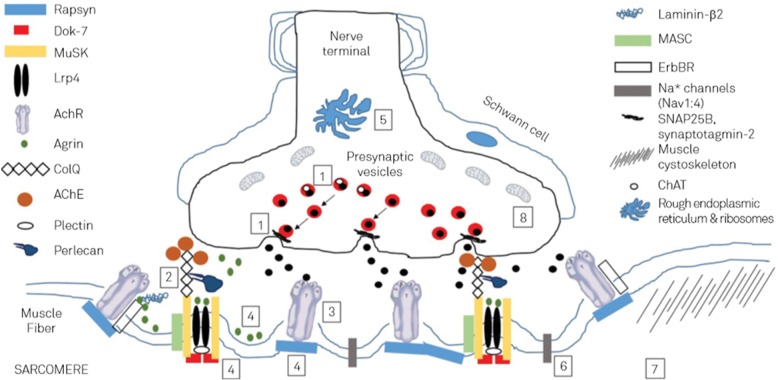
Results: Currently, mutations in 32 genes are made responsible for autosomal dominant or autosomal recessive CMSs. These mutations concern 8 presynaptic, 4 synaptic, 15 post-synaptic, and 5 glycosilation proteins. These proteins function as ion-channels, enzymes, or structural, signalling, sensor, or transporter proteins. The most common causative genes are CHAT, COLQ, RAPSN, CHRNE, DOK7, and GFPT1. Phenotypically, these mutations manifest as abnormal fatigability or permanent or fluctuating weakness of extra-ocular, facial, bulbar, axial, respiratory, or limb muscles, hypotonia, or developmental delay. Cognitive disability, dysmorphism, neuropathy, or epilepsy are rare. Low- or high-frequency repetitive nerve stimulation may show an abnormal increment or decrement, and SF-EMG an increased jitter or blockings. Most CMSs respond favourably to acetylcholine-esterase inhibitors, 3,4-diamino-pyridine, salbutamol, albuterol, ephedrine, fluoxetine, or atracurium.
Conclusions: CMSs are an increasingly recognised group of genetically transmitted defects, which usually respond favorably to drugs enhancing the neuromuscular transmission. CMSs need to be differentiated from neuromuscular disorders due to muscle or nerve dysfunction.
Keywords: Fatigue; Genes; Hereditary; Mutation; Myasthenia; Myasthenic syndrome; Repetitive nerve stimulation; Weakness.
Conflict of interest statement
Ethics approval and consent to participate
not applicable.
Consent for publication
not applicable.
Competing interests
the author declares that he has no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Bestue-Cardiel M, Natera-de BD. Current status of congenital myasthenic syndromes. Rev Neurol. 2017;65:161–176. - PubMed
-
- Gomez CM, Gammack JT. A leucine-to-phenylalanine substitution in the acetylcholine receptor ion channel in a family with the slow-channel syndrome. Neurology. 1995;45:982–985. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
