

Insights Into an Unexplored Component of the Mosquito Repeatome: Distribution and Variability of Viral Sequences Integrated Into the Genome of the Arboviral Vector Aedes albopictus
- PMID: 30809249
- PMCID: PMC6379468
- DOI: 10.3389/fgene.2019.00093
Insights Into an Unexplored Component of the Mosquito Repeatome: Distribution and Variability of Viral Sequences Integrated Into the Genome of the Arboviral Vector Aedes albopictus
Abstract
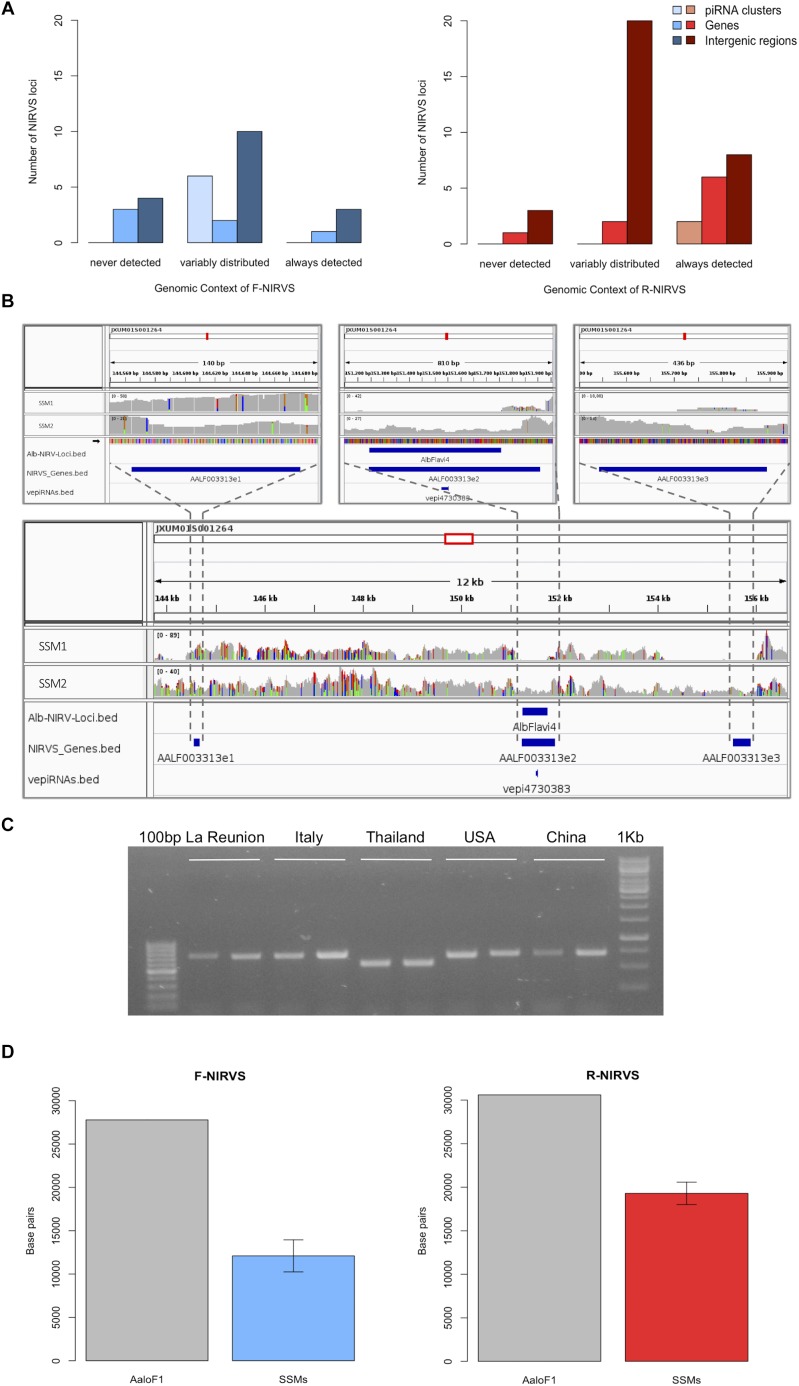
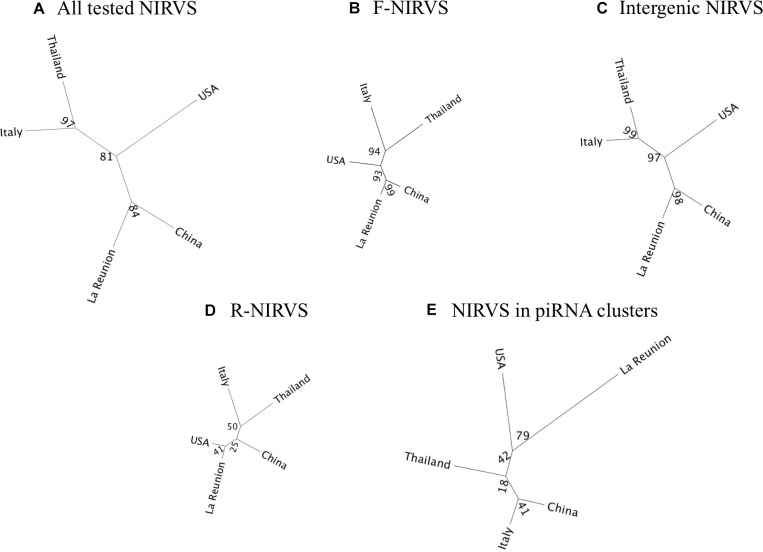
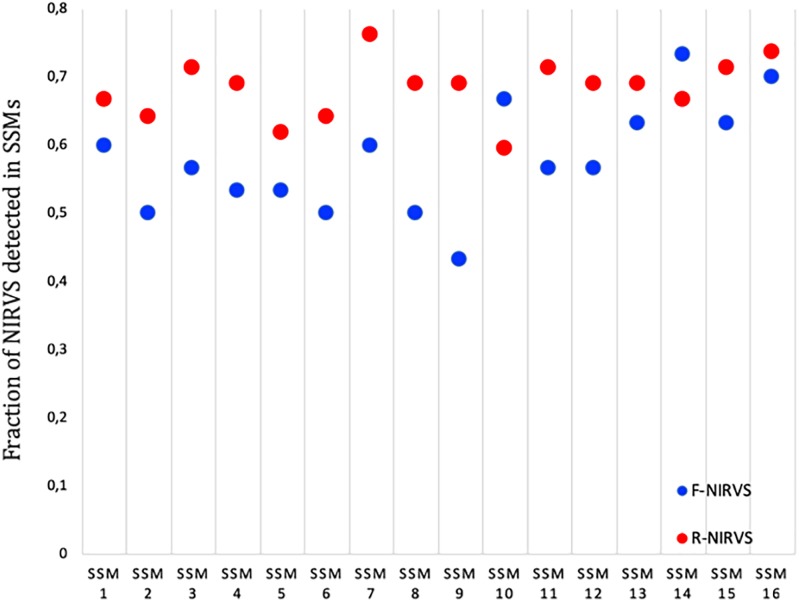
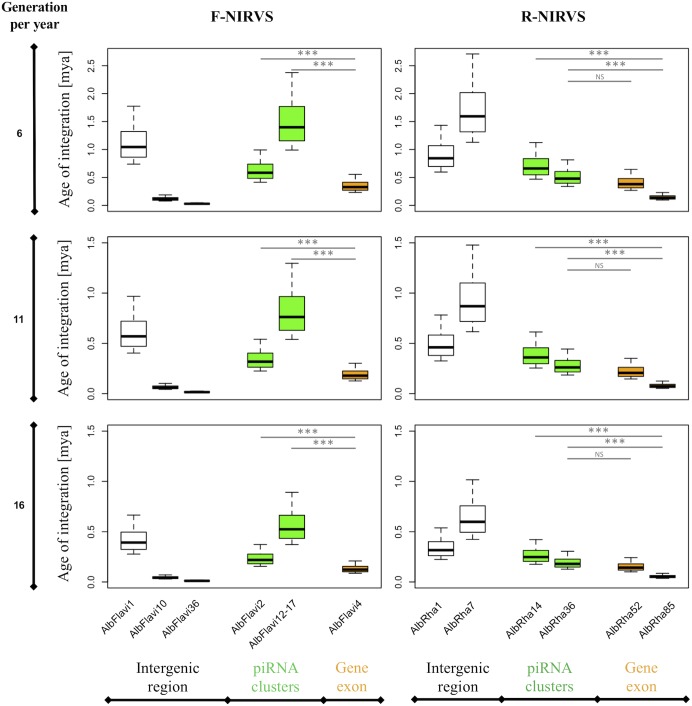
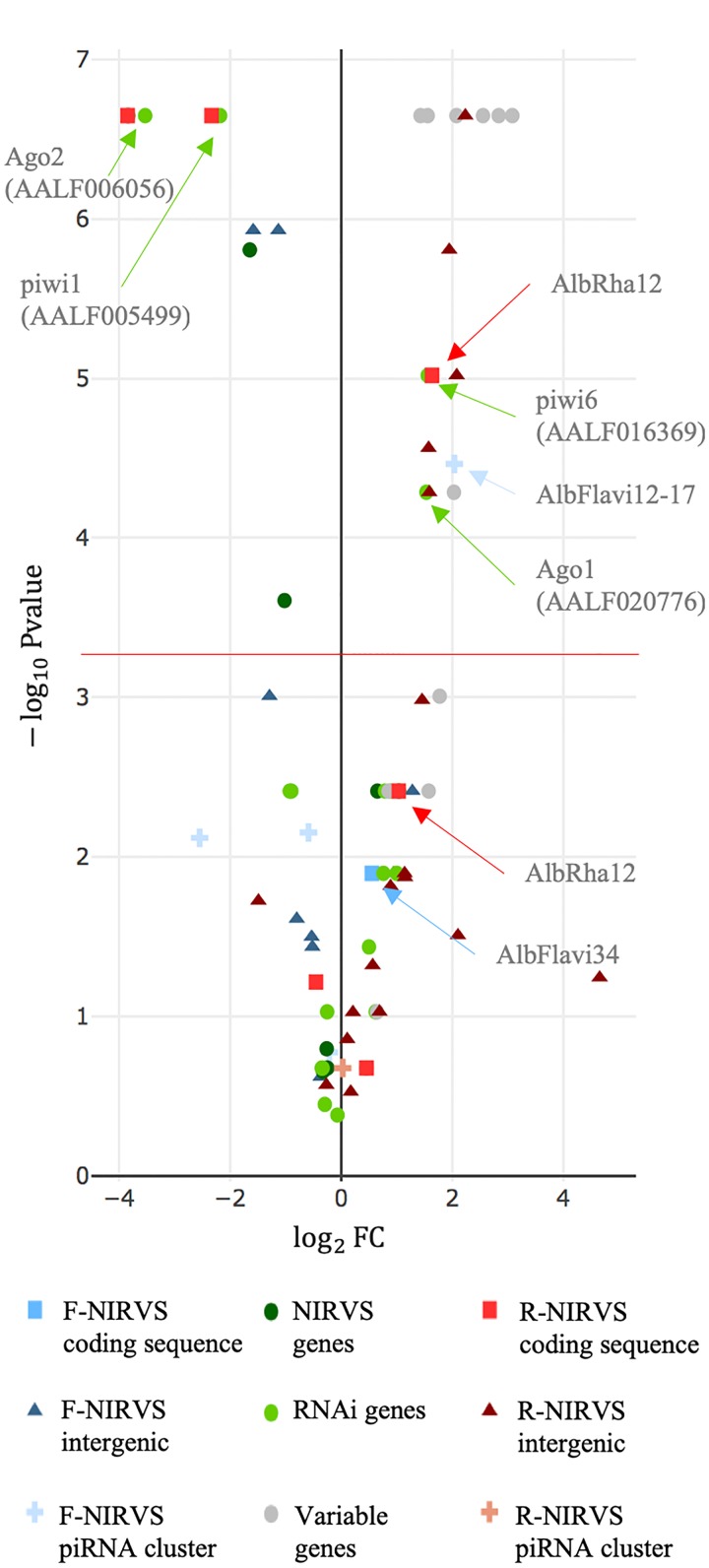
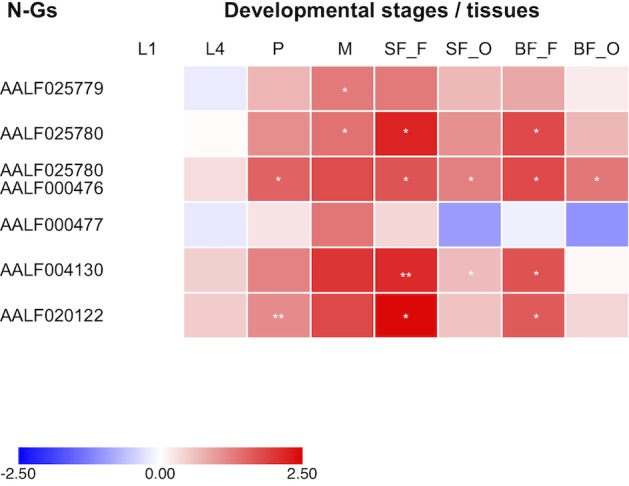
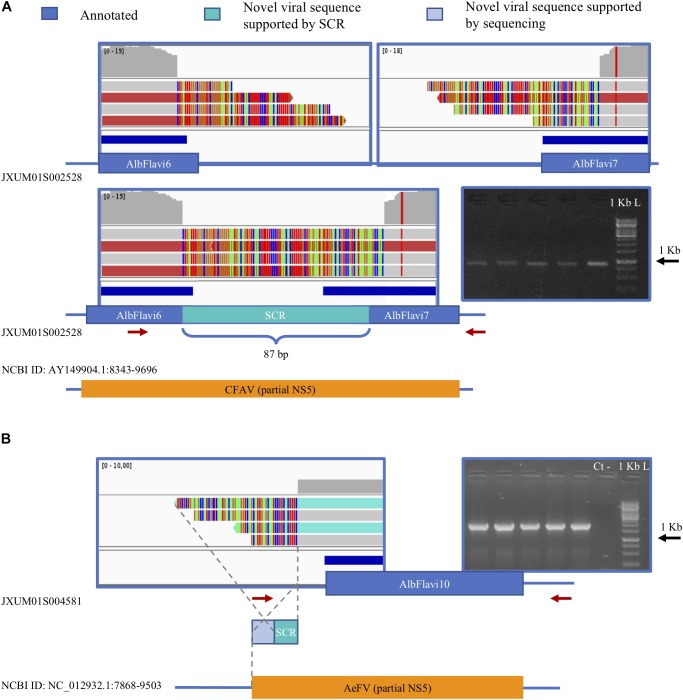
The Asian tiger mosquito Aedes albopictus is an invasive mosquito and a competent vector for public-health relevant arboviruses such as Chikungunya (Alphavirus), Dengue and Zika (Flavivirus) viruses. Unexpectedly, the sequencing of the genome of this mosquito revealed an unusually high number of integrated sequences with similarities to non-retroviral RNA viruses of the Flavivirus and Rhabdovirus genera. These Non-retroviral Integrated RNA Virus Sequences (NIRVS) are enriched in piRNA clusters and coding sequences and have been proposed to constitute novel mosquito immune factors. However, given the abundance of NIRVS and their variable viral origin, their relative biological roles remain unexplored. Here we used an analytical approach that intersects computational, evolutionary and molecular methods to study the genomic landscape of mosquito NIRVS. We demonstrate that NIRVS are differentially distributed across mosquito genomes, with a core set of seemingly the oldest integrations with similarity to Rhabdoviruses. Additionally, we compare the polymorphisms of NIRVS with respect to that of fast and slow-evolving genes within the Ae. albopictus genome. Overall, NIRVS appear to be less polymorphic than slow-evolving genes, with differences depending on whether they occur in intergenic regions or in piRNA clusters. Finally, two NIRVS that map within the coding sequences of genes annotated as Rhabdovirus RNA-dependent RNA polymerase and the nucleocapsid-encoding gene, respectively, are highly polymorphic and are expressed, suggesting exaptation possibly to enhance the mosquito's antiviral responses. These results greatly advance our understanding of the complexity of the mosquito repeatome and the biology of viral integrations in mosquito genomes.
Keywords: domestication; immunity; mosquitoes; piRNA pathway; repeatome; viral integrations.
Figures
Similar articles
-
Evolution and biological significance of flaviviral elements in the genome of the arboviral vector Aedes albopictus.Emerg Microbes Infect. 2019;8(1):1265-1279. doi: 10.1080/22221751.2019.1657785. Emerg Microbes Infect. 2019. PMID: 31469046 Free PMC article.
-
Comparative genomics shows that viral integrations are abundant and express piRNAs in the arboviral vectors Aedes aegypti and Aedes albopictus.BMC Genomics. 2017 Jul 5;18(1):512. doi: 10.1186/s12864-017-3903-3. BMC Genomics. 2017. PMID: 28676109 Free PMC article.
-
Endogenous non-retroviral elements in genomes of Aedes mosquitoes and vector competence.Emerg Microbes Infect. 2019;8(1):542-555. doi: 10.1080/22221751.2019.1599302. Emerg Microbes Infect. 2019. PMID: 30938223 Free PMC article. Review.
-
Novel insights into endogenous RNA viral elements in Ixodes scapularis and other arbovirus vector genomes.Virus Evol. 2019 Jun 18;5(1):vez010. doi: 10.1093/ve/vez010. eCollection 2019 Jan. Virus Evol. 2019. PMID: 31249694 Free PMC article.
-
The Widespread Occurrence and Potential Biological Roles of Endogenous Viral Elements in Insect Genomes.Curr Issues Mol Biol. 2020;34:13-30. doi: 10.21775/cimb.034.013. Epub 2019 Jun 6. Curr Issues Mol Biol. 2020. PMID: 31167954 Review.
Cited by
-
Polymorphism analyses and protein modelling inform on functional specialization of Piwi clade genes in the arboviral vector Aedes albopictus.PLoS Negl Trop Dis. 2019 Dec 2;13(12):e0007919. doi: 10.1371/journal.pntd.0007919. eCollection 2019 Dec. PLoS Negl Trop Dis. 2019. PMID: 31790401 Free PMC article.
-
In and Outs of Chuviridae Endogenous Viral Elements: Origin of a Potentially New Retrovirus and Signature of Ancient and Ongoing Arms Race in Mosquito Genomes.Front Genet. 2020 Oct 22;11:542437. doi: 10.3389/fgene.2020.542437. eCollection 2020. Front Genet. 2020. PMID: 33193616 Free PMC article.
-
A Novel Anphevirus in Aedes albopictus Mosquitoes Is Distributed Worldwide and Interacts with the Host RNA Interference Pathway.Viruses. 2020 Nov 6;12(11):1264. doi: 10.3390/v12111264. Viruses. 2020. PMID: 33172032 Free PMC article.
-
Evolution and biological significance of flaviviral elements in the genome of the arboviral vector Aedes albopictus.Emerg Microbes Infect. 2019;8(1):1265-1279. doi: 10.1080/22221751.2019.1657785. Emerg Microbes Infect. 2019. PMID: 31469046 Free PMC article.
-
Arboviruses and the Challenge to Establish Systemic and Persistent Infections in Competent Mosquito Vectors: The Interaction With the RNAi Mechanism.Front Physiol. 2019 Jul 11;10:890. doi: 10.3389/fphys.2019.00890. eCollection 2019. Front Physiol. 2019. PMID: 31354527 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources