

Personalized medicine in chronic kidney disease by detection of monogenic mutations
- PMID: 30809662
- PMCID: PMC7057531
- DOI: 10.1093/ndt/gfz028
Personalized medicine in chronic kidney disease by detection of monogenic mutations
Abstract
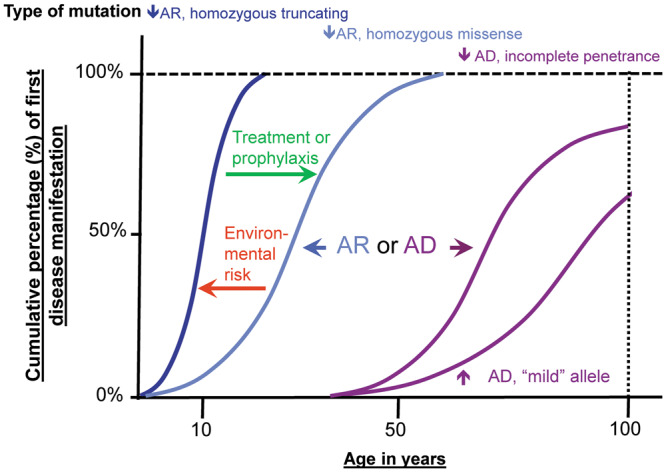
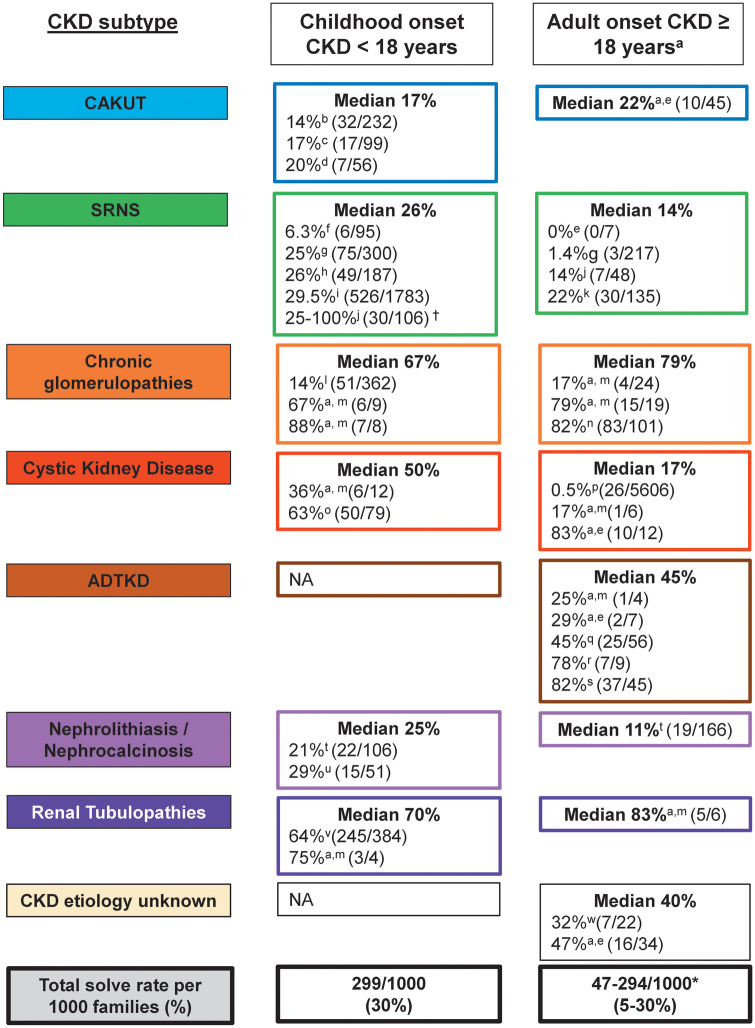
A large fraction of early-onset chronic kidney disease (CKD) is known to be monogenic in origin. To date, ∼450 monogenic (synonymous with single-gene disorders) genes, if mutated, are known to cause CKD, explaining ∼30% of cases in pediatric cohorts and ∼5-30% in adult cohorts. However, there are likely hundreds of additional monogenic nephropathy genes that may be revealed by whole-exome or -genome sequencing. Although the discovery of novel CKD-causing genes has accelerated, significant challenges in adult populations remain due to broad phenotypic heterogeneity together with variable expressivity, incomplete penetrance or age-related penetrance of these genes. Here we give an overview of the currently known monogenic causes for human CKD. We also describe how next-generation sequencing facilitates rapid molecular genetic diagnostics in individuals with suspected genetic kidney disease. In an era of precision medicine, understanding the utility of genetic testing in individuals with a suspected inherited nephropathy has important diagnostic and prognostic implications. Detection of monogenic causes of CKD permits molecular genetic diagnosis for patients and families and opens avenues for personalized treatment strategies for CKD. As an example, detection of a pathogenic mutation in the gene HNF1B not only allows for the formal diagnosis of CKD, but can also facilitate screening for additional extrarenal manifestations of disease, such as maturity-onset diabetes of youth, subclinical abnormal liver function tests, neonatal cholestasis and pancreatic hypoplasia. It also provides the driving force towards a better understanding of disease pathogenesis, potentially facilitating targeted new therapies for individuals with CKD.
Keywords: Mendelian renal disease; familial nephropathy; genetic kidney disease; monogenic disease; single-gene disorders.
© The Author(s) 2019. Published by Oxford University Press on behalf of ERA-EDTA. All rights reserved.
Figures

References
-
- Inker LA, Astor BC, Fox CH et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. Am J Kidney Dis 2014; 63: 713–735 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
