

Inflammation and Liver Cancer: Molecular Mechanisms and Therapeutic Targets
- PMID: 30809789
- PMCID: PMC6616367
- DOI: 10.1055/s-0038-1676806
Inflammation and Liver Cancer: Molecular Mechanisms and Therapeutic Targets
Abstract
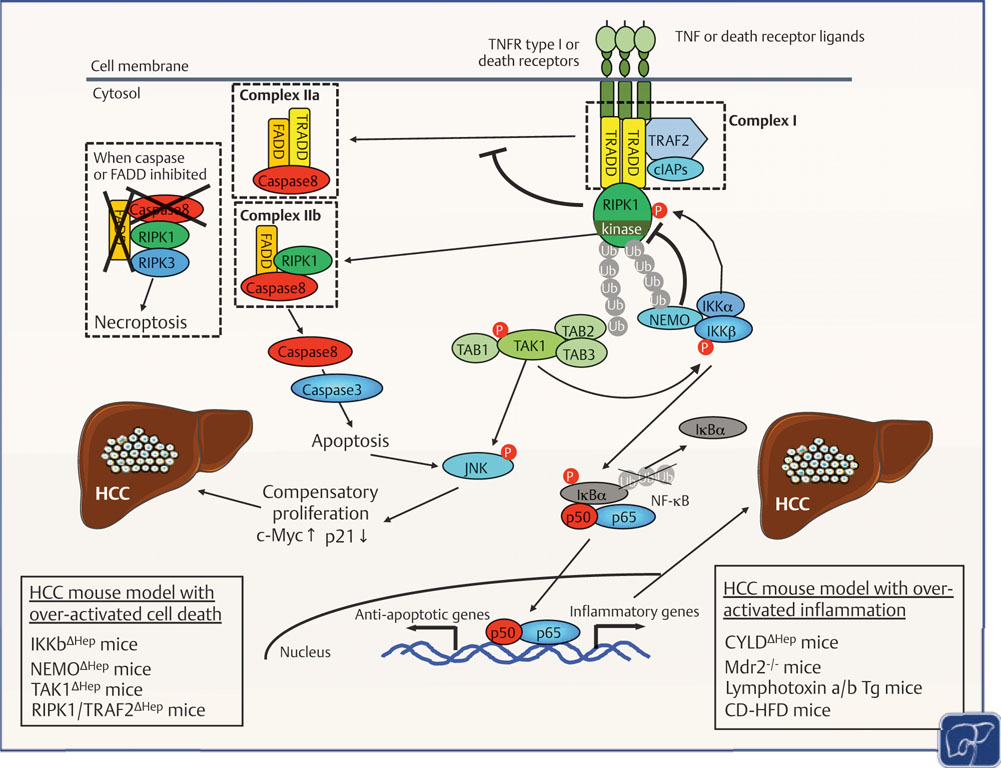
Hepatocellular carcinoma (HCC) is associated with chronic inflammation and fibrosis arising from different etiologies, including hepatitis B and C and alcoholic and nonalcoholic fatty liver diseases. The inflammatory cytokines tumor necrosis factor-α and interleukin-6 and their downstream targets nuclear factor kappa B (NF-κB), c-Jun N-terminal kinase (JNK), and signal transducer and activator of transcription 3 drive inflammation-associated HCC. Further, while adaptive immunity promotes immune surveillance to eradicate early HCC, adaptive immune cells, such as CD8+ T cells, Th17 cells, and B cells, can also stimulate HCC development. Thus, the role of the hepatic immune system in HCC development is a highly complex topic. This review highlights the role of cytokine signals, NF-κB, JNK, innate and adaptive immunity, and hepatic stellate cells in HCC and discusses whether these pathways could be therapeutic targets. The authors will also discuss cholangiocarcinoma and liver metastasis because biliary inflammation and tumor-associated stroma are essential for cholangiocarcinoma development and because primary tumor-derived inflammatory mediators promote the formation of a "premetastasis niche" in the liver.
Thieme Medical Publishers 333 Seventh Avenue, New York, NY 10001, USA.
Conflict of interest statement
Disclosure The authors report no conflicts of interest in this work.
Figures



References
-
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin 2018;68(01):7–30 - PubMed
-
- El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007;132(07):2557–2576 - PubMed
-
- Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013;10(11):656–665 - PubMed
-
- Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol 2016;13(02):88–110 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
