

Regulation of IL-17A and implications for TGF-β1 comodulation of airway smooth muscle remodeling in severe asthma
- PMID: 30810068
- PMCID: PMC6589583
- DOI: 10.1152/ajplung.00416.2018
Regulation of IL-17A and implications for TGF-β1 comodulation of airway smooth muscle remodeling in severe asthma
Abstract
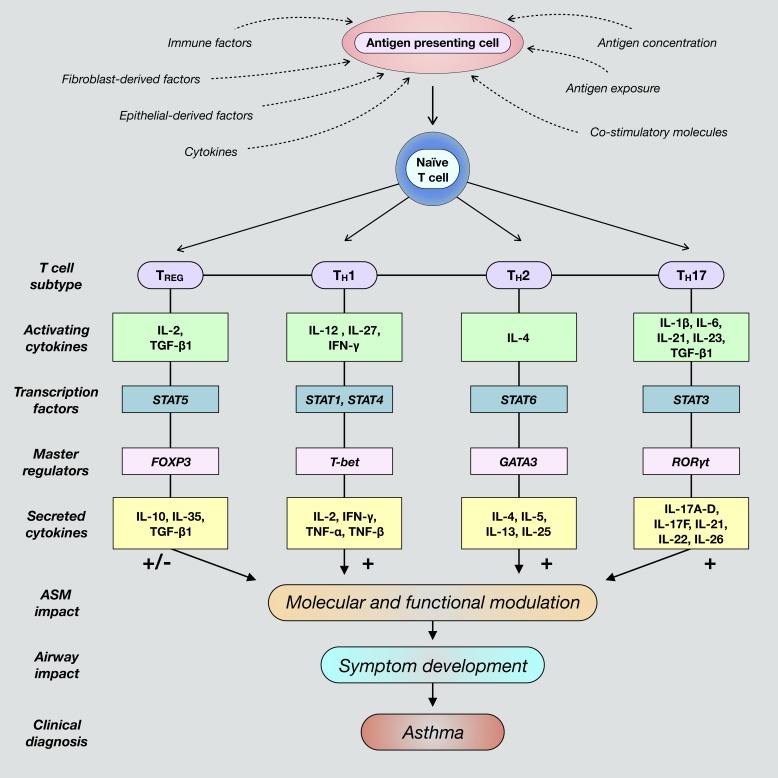
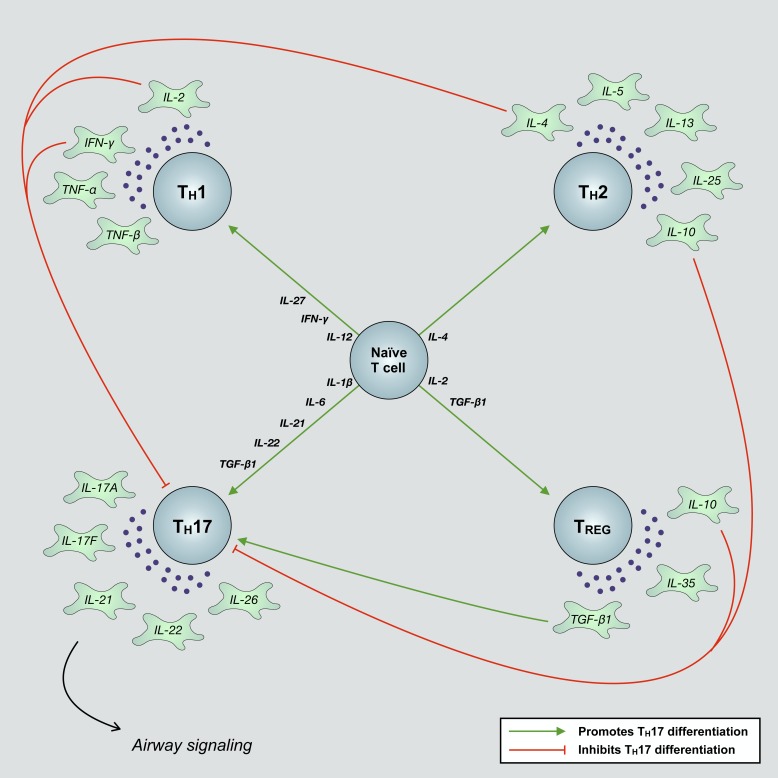
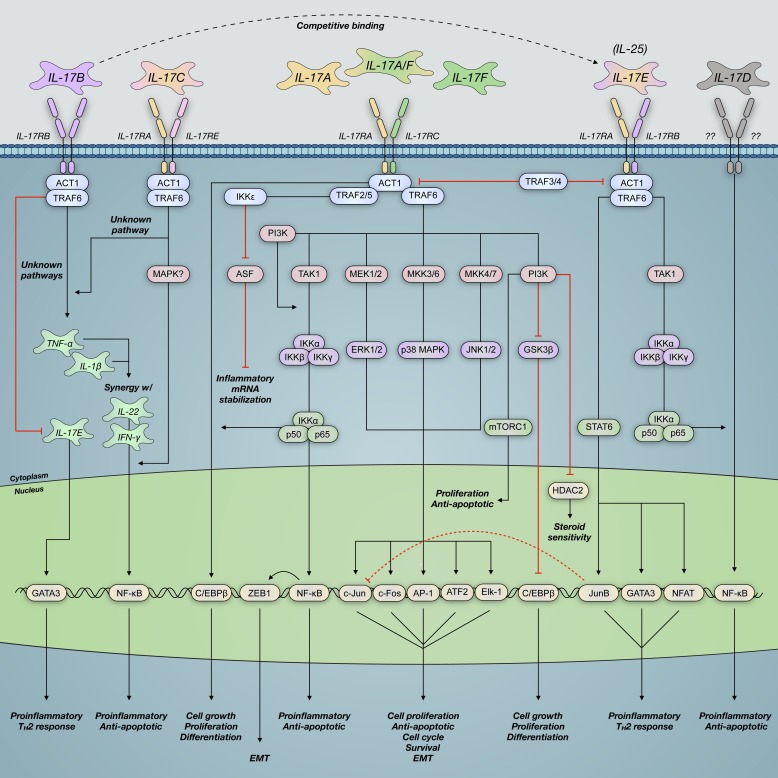
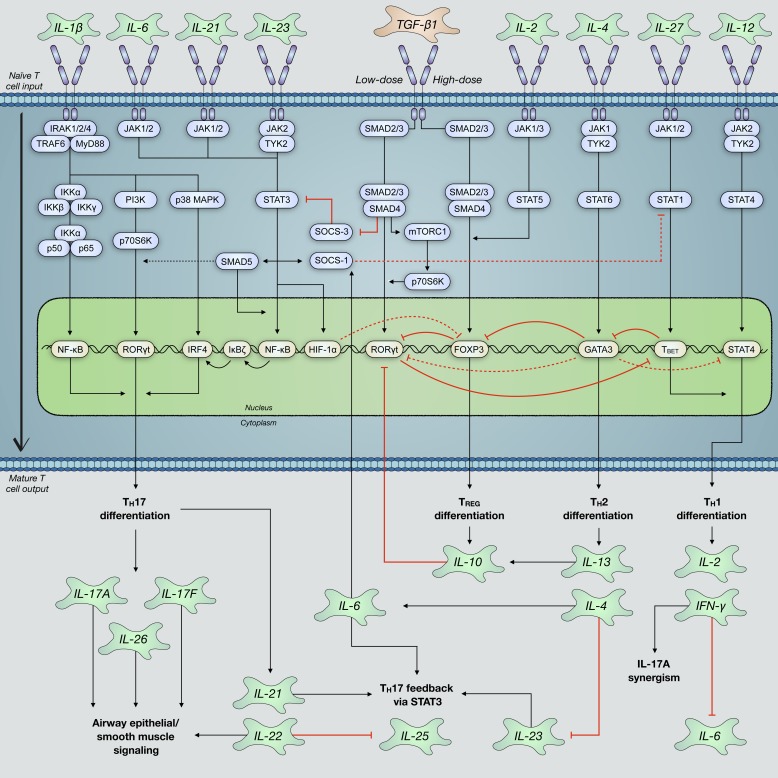
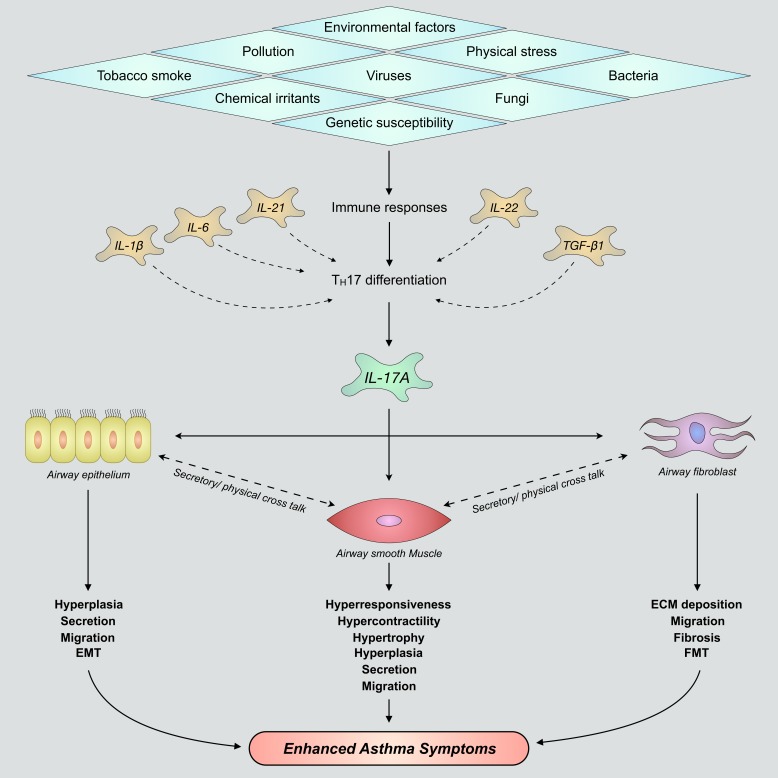
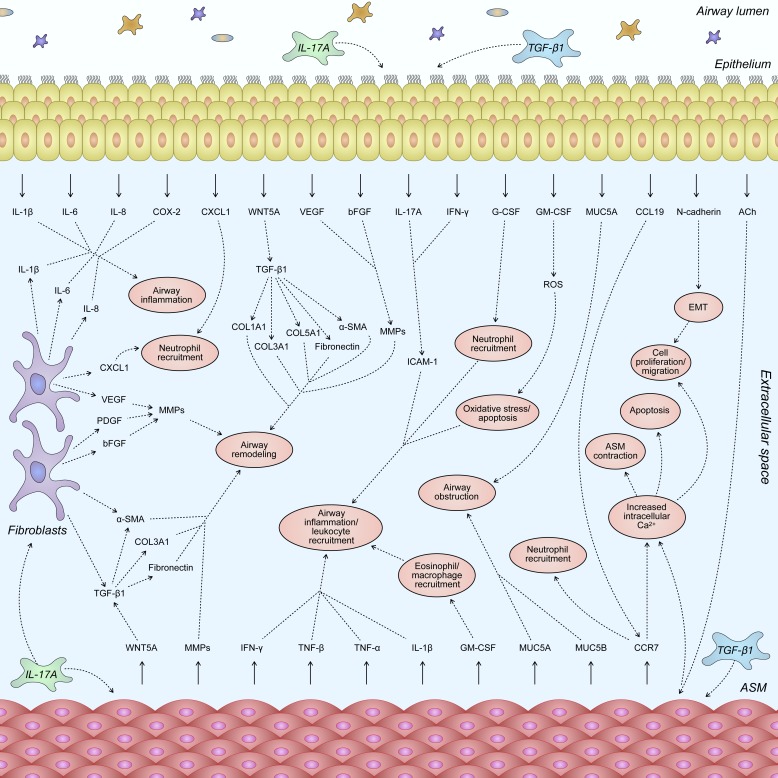
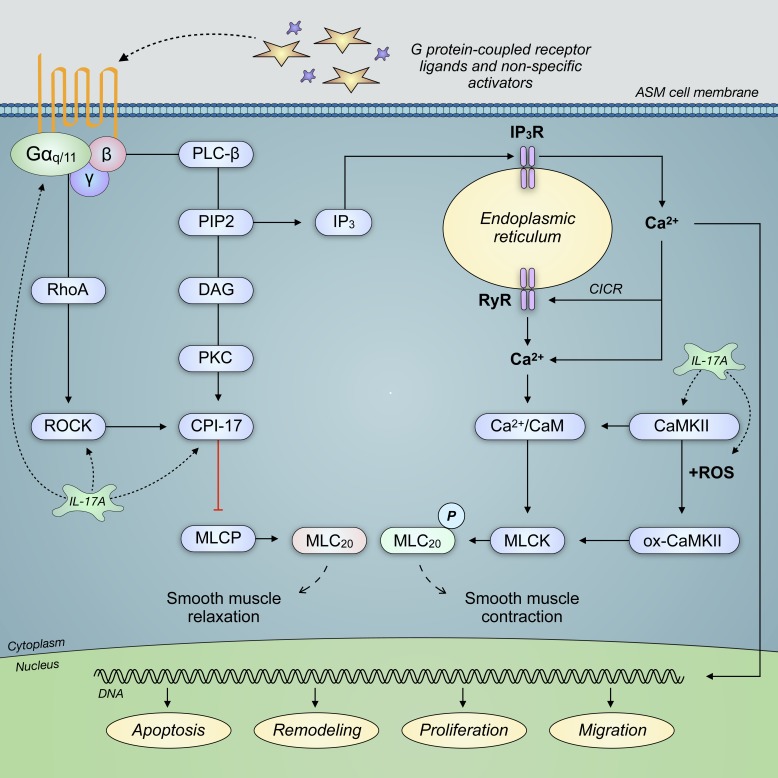
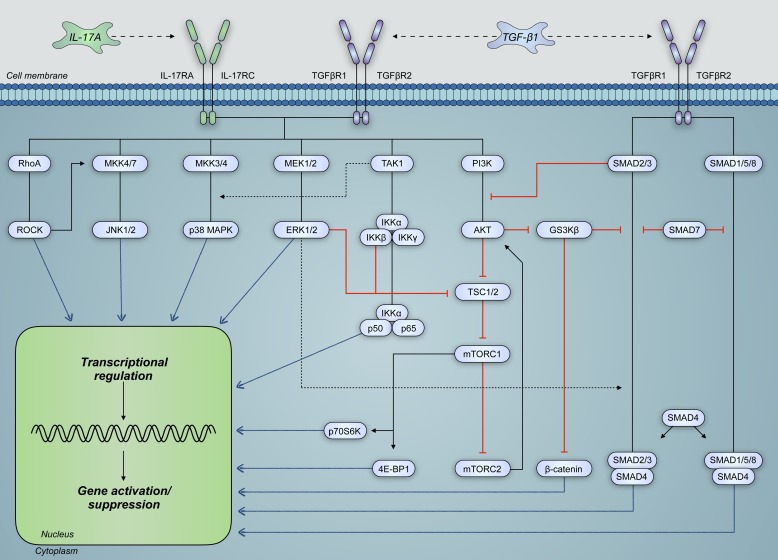
Severe asthma develops as a result of heightened, persistent symptoms that generally coincide with pronounced neutrophilic airway inflammation. In individuals with severe asthma, symptoms are poorly controlled by high-dose inhaled glucocorticoids and often lead to elevated morbidity and mortality rates that underscore the necessity for novel drug target identification that overcomes limitations in disease management. Many incidences of severe asthma are mechanistically associated with T helper 17 (TH17) cell-derived cytokines and immune factors that mediate neutrophilic influx to the airways. TH17-secreted interleukin-17A (IL-17A) is an independent risk factor for severe asthma that impacts airway smooth muscle (ASM) remodeling. TH17-derived cytokines and diverse immune mediators further interact with structural cells of the airway to induce pathophysiological processes that impact ASM functionality. Transforming growth factor-β1 (TGF-β1) is a pivotal mediator involved in airway remodeling that correlates with enhanced TH17 activity in individuals with severe asthma and is essential to TH17 differentiation and IL-17A production. IL-17A can also reciprocally enhance activation of TGF-β1 signaling pathways, whereas combined TH1/TH17 or TH2/TH17 immune responses may additively impact asthma severity. This review seeks to provide a comprehensive summary of cytokine-driven T cell fate determination and TH17-mediated airway inflammation. It will further review the evidence demonstrating the extent to which IL-17A interacts with various immune factors, specifically TGF-β1, to contribute to ASM remodeling and altered function in TH17-driven endotypes of severe asthma.
Keywords: IL-17A; TGF-β1; airway remodeling; airway smooth muscle; asthma.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
References
-
- Alrashdan YA, Alkhouri H, Chen E, Lalor DJ, Poniris M, Henness S, Brightling CE, Burgess JK, Armour CL, Ammit AJ, Hughes JM. Asthmatic airway smooth muscle CXCL10 production: mitogen-activated protein kinase JNK involvement. Am J Physiol Lung Cell Mol Physiol 302: L1118–L1127, 2012. doi:10.1152/ajplung.00232.2011. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous