

NADPH Oxidase Mediates Membrane Androgen Receptor-Induced Neurodegeneration
- PMID: 30811529
- PMCID: PMC6435014
- DOI: 10.1210/en.2018-01079
NADPH Oxidase Mediates Membrane Androgen Receptor-Induced Neurodegeneration
Abstract
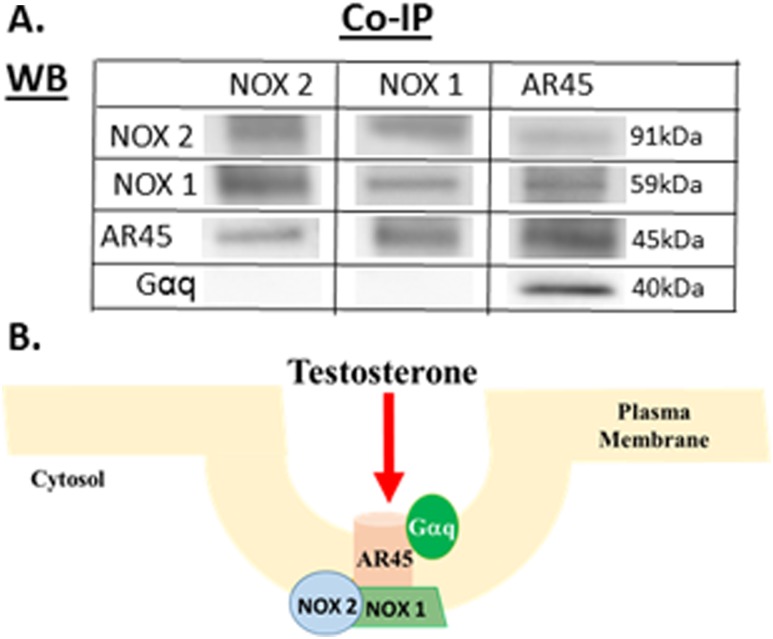
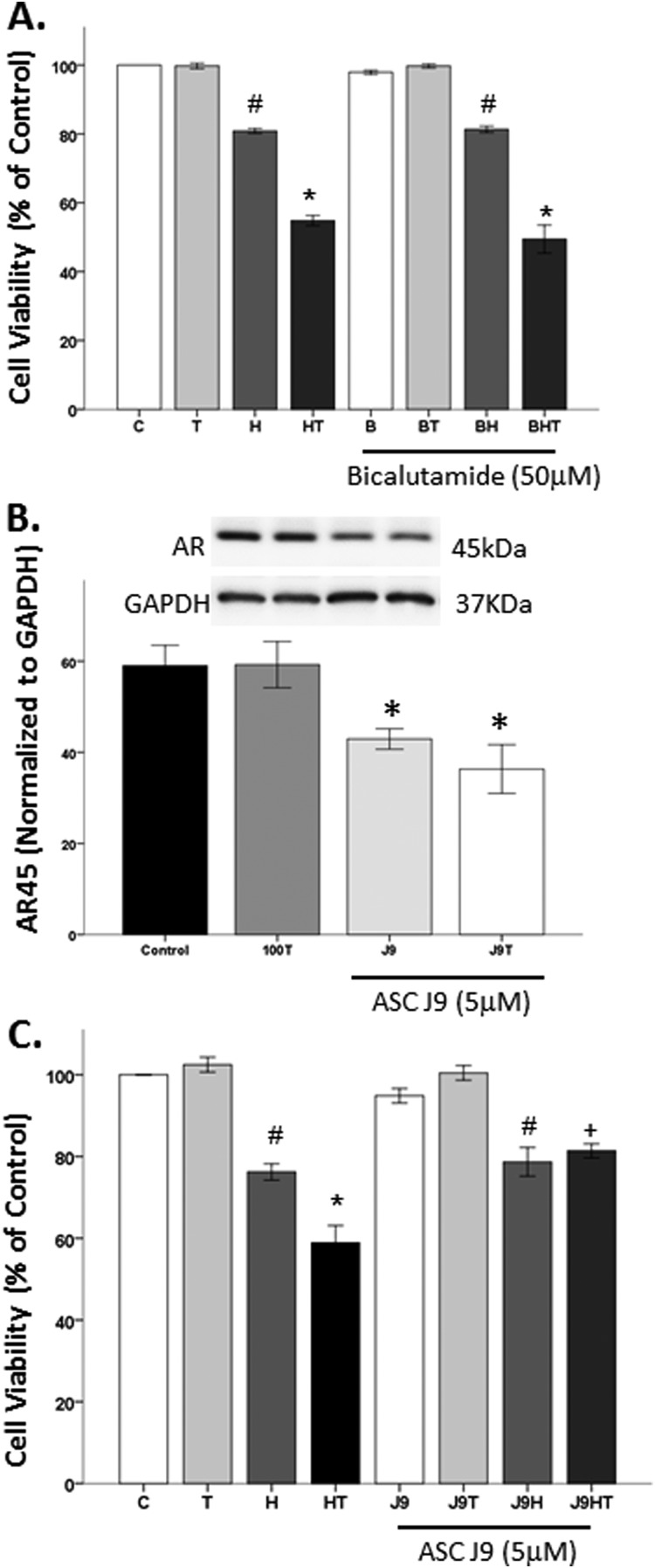
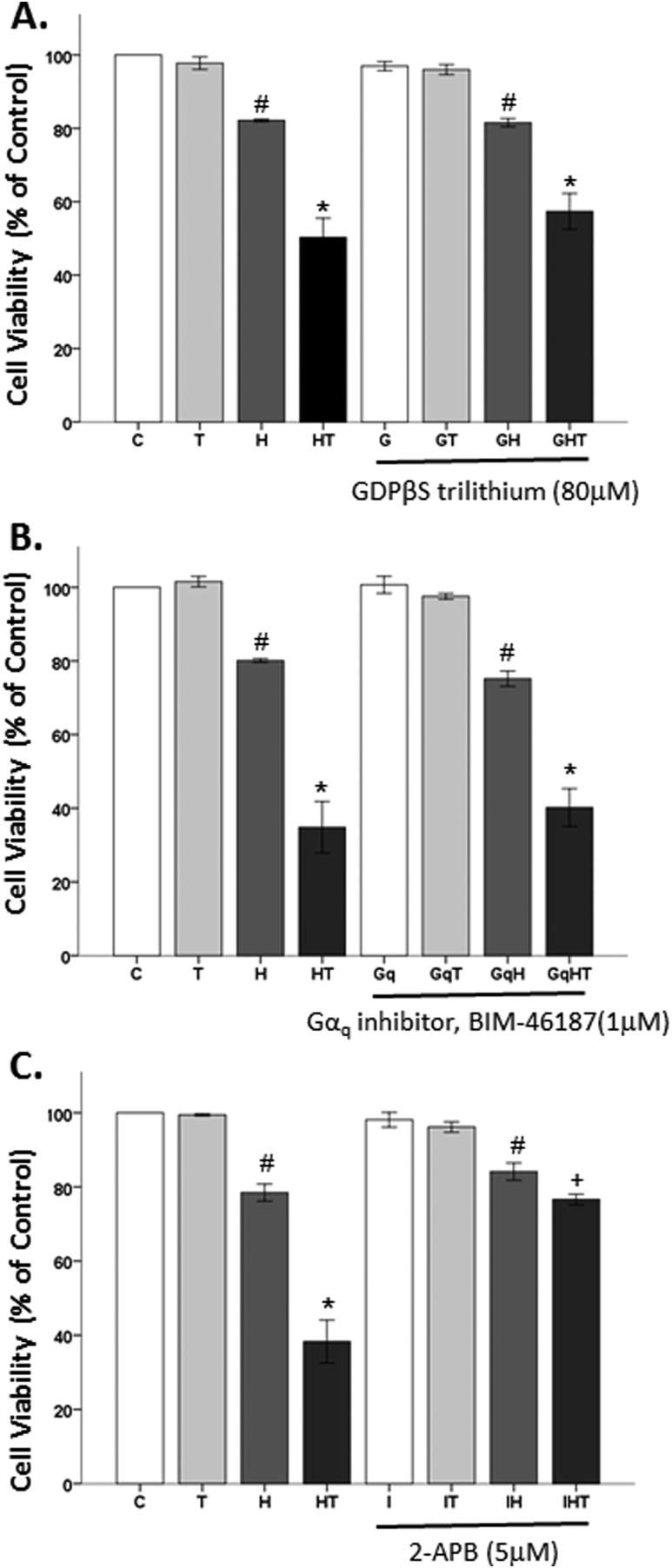
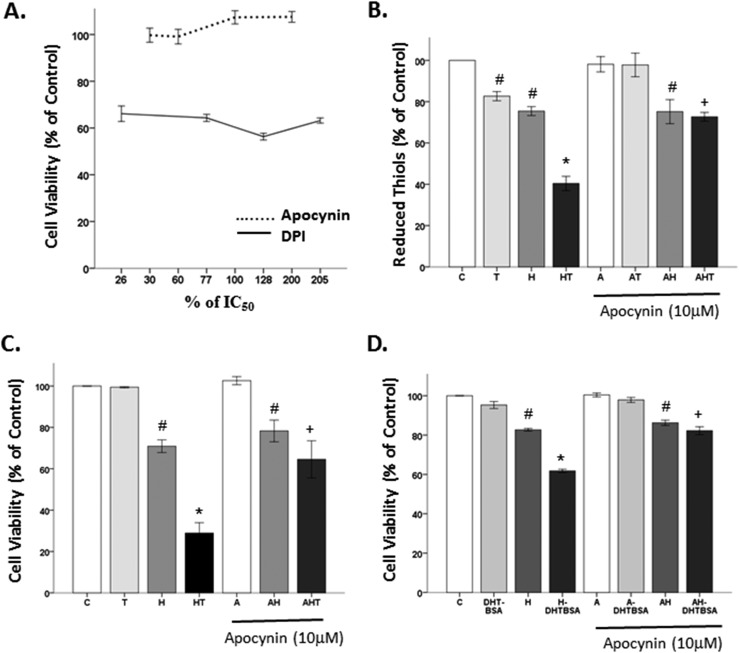
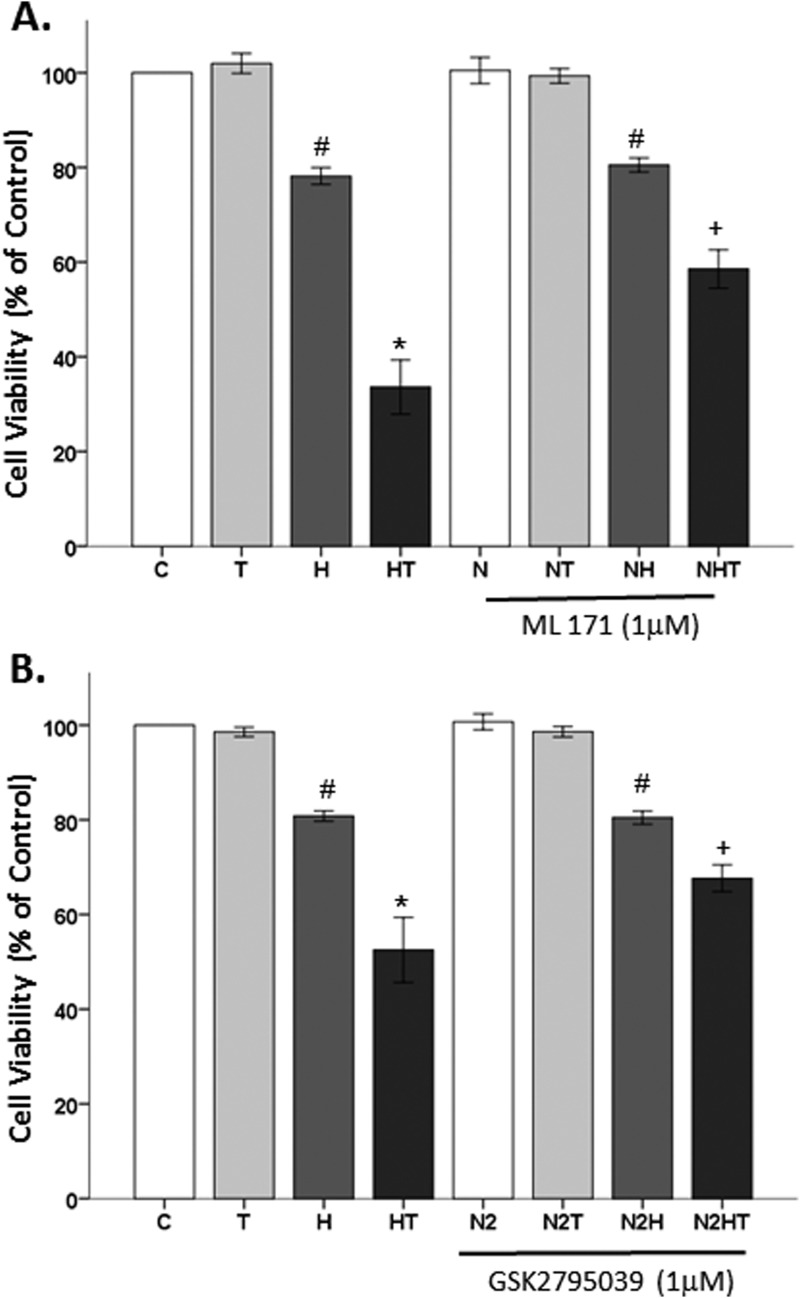
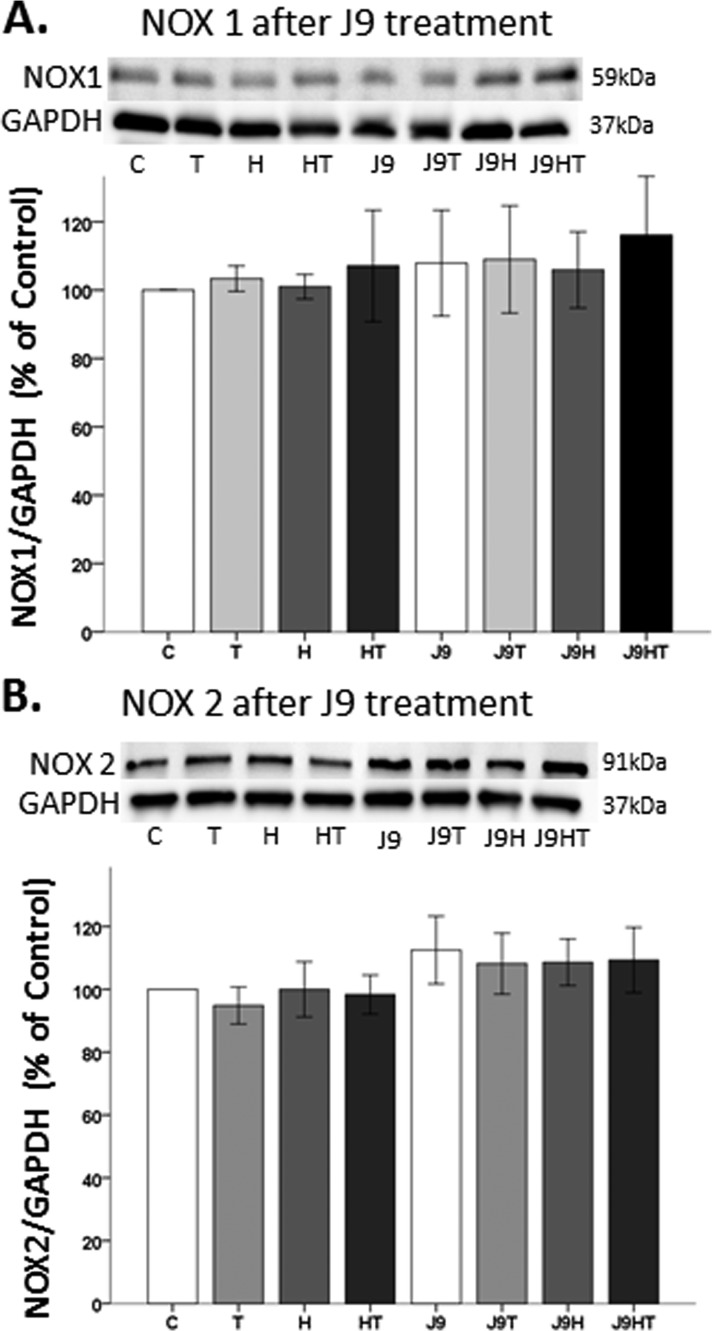
Oxidative stress (OS) is a common characteristic of several neurodegenerative disorders, including Parkinson disease (PD). PD is more prevalent in men than in women, indicating the possible involvement of androgens. Androgens can have either neuroprotective or neurodamaging effects, depending on the presence of OS. Specifically, in an OS environment, androgens via a membrane-associated androgen receptor (mAR) exacerbate OS-induced damage. To investigate the role of androgens on OS signaling and neurodegeneration, the effects of testosterone and androgen receptor activation on the major OS signaling cascades, the reduced form of NAD phosphate (NADPH) oxidase (NOX)1 and NOX2 and the Gαq/inositol trisphosphate receptor (InsP3R), were examined. To create an OS environment, an immortalized neuronal cell line was exposed to H2O2 prior to cell-permeable/cell-impermeable androgens. Different inhibitors were used to examine the role of G proteins, mAR, InsP3R, and NOX1/2 on OS generation and cell viability. Both testosterone and DHT/3-O-carboxymethyloxime (DHT)-BSA increased H2O2-induced OS and cell death, indicating the involvement of an mAR. Furthermore, classical AR antagonists did not block testosterone's negative effects in an OS environment. Because there are no known antagonists specific for mARs, an AR protein degrader, ASC-J9, was used to block mAR action. ASC-J9 blocked testosterone's negative effects. To determine OS-related signaling mediated by mAR, this study examined NOX1, NOX2, Gαq. NOX1, NOX2, and the Gαq complex with mAR. Only NOX inhibition blocked testosterone-induced cell loss and OS. No effects of blocking either Gαq or G protein activation were observed on testosterone's negative effects. These results indicate that androgen-induced OS is via the mAR-NOX complex and not the mAR-Gαq complex.
Copyright © 2019 Endocrine Society.
Figures

Similar articles
-
Androgen stimulates endothelial cell proliferation via an androgen receptor/VEGF/cyclin A-mediated mechanism.Am J Physiol Heart Circ Physiol. 2011 Apr;300(4):H1210-21. doi: 10.1152/ajpheart.01210.2010. Epub 2011 Jan 21. Am J Physiol Heart Circ Physiol. 2011. PMID: 21257919 Free PMC article.
-
Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor-mediated pathway.Endocrinology. 2003 Nov;144(11):5081-8. doi: 10.1210/en.2003-0741. Epub 2003 Jul 24. Endocrinology. 2003. PMID: 12960001
-
Activation of a membrane-associated androgen receptor promotes cell death in primary cortical astrocytes.Endocrinology. 2007 May;148(5):2458-64. doi: 10.1210/en.2006-1443. Epub 2007 Feb 15. Endocrinology. 2007. PMID: 17303658
-
Prolactin influences upon androgen action in male accessory sex organs.Adv Sex Horm Res. 1976;2:425-70. Adv Sex Horm Res. 1976. PMID: 189591 Review.
-
Membrane Androgen Receptors Unrelated to Nuclear Steroid Receptors.Endocrinology. 2019 Apr 1;160(4):772-781. doi: 10.1210/en.2018-00987. Endocrinology. 2019. PMID: 30753403 Review.
Cited by
-
Sex and age differences in social and cognitive function in offspring exposed to late gestational hypoxia.Biol Sex Differ. 2023 Nov 11;14(1):81. doi: 10.1186/s13293-023-00557-0. Biol Sex Differ. 2023. PMID: 37951901 Free PMC article.
-
Chronic intermittent hypoxia-induced hypertension: the impact of sex hormones.Am J Physiol Regul Integr Comp Physiol. 2024 May 1;326(5):R333-R345. doi: 10.1152/ajpregu.00258.2023. Epub 2024 Feb 26. Am J Physiol Regul Integr Comp Physiol. 2024. PMID: 38406843 Free PMC article. Review.
-
Neuroprotective and neurotoxic outcomes of androgens and estrogens in an oxidative stress environment.Biol Sex Differ. 2020 Mar 29;11(1):12. doi: 10.1186/s13293-020-0283-1. Biol Sex Differ. 2020. PMID: 32223745 Free PMC article.
-
The impact of chronic intermittent hypoxia on enzymatic activity in memory-associated brain regions of male and female rats.Res Sq [Preprint]. 2024 Dec 13:rs.3.rs-5449794. doi: 10.21203/rs.3.rs-5449794/v1. Res Sq. 2024. Update in: Biol Sex Differ. 2025 Jan 31;16(1):5. doi: 10.1186/s13293-025-00688-6. PMID: 39711575 Free PMC article. Updated. Preprint.
-
The Role of Lipid Rafts and Membrane Androgen Receptors in Androgen's Neurotoxic Effects.J Endocr Soc. 2022 Feb 21;6(5):bvac030. doi: 10.1210/jendso/bvac030. eCollection 2022 May 1. J Endocr Soc. 2022. PMID: 35308305 Free PMC article.
References
-
- Sherer TB, Chowdhury S, Peabody K, Brooks DW. Overcoming obstacles in Parkinson’s disease. Mov Disord. 2012;27(13):1606–1611. - PubMed
-
- Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68(5):384–386. - PubMed
-
- Ross GW, Petrovitch H, Abbott RD, Nelson J, Markesbery W, Davis D, Hardman J, Launer L, Masaki K, Tanner CM, White LR. Parkinsonian signs and substantia nigra neuron density in decendents elders without PD. Ann Neurol. 2004;56(4):532–539. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
