

Novel SCN2A mutation in a family associated with juvenile-onset myoclonus: Case report
- PMID: 30813219
- PMCID: PMC6408085
- DOI: 10.1097/MD.0000000000014698
Novel SCN2A mutation in a family associated with juvenile-onset myoclonus: Case report
Abstract
Rationale: The phenotypic spectrum caused by SCN2A mutations includes benign neonatal/infantile seizures, Ohtahara syndrome, infantile spasms, West syndrome, and other unclassified epileptic phenotypes. Mutations in SCN2A have been implicated in neonatal seizure cases. Here, we described a Chinese family with 2 members having juvenile-onset myoclonus and identified a novel SCN2A point mutation within this family.
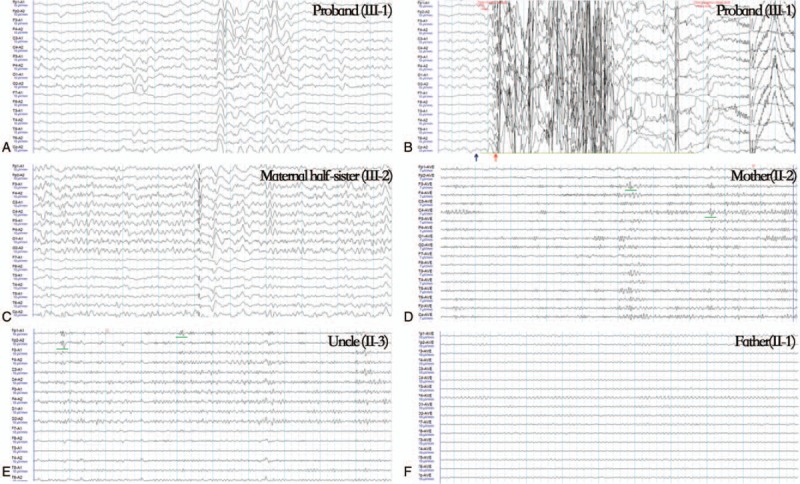
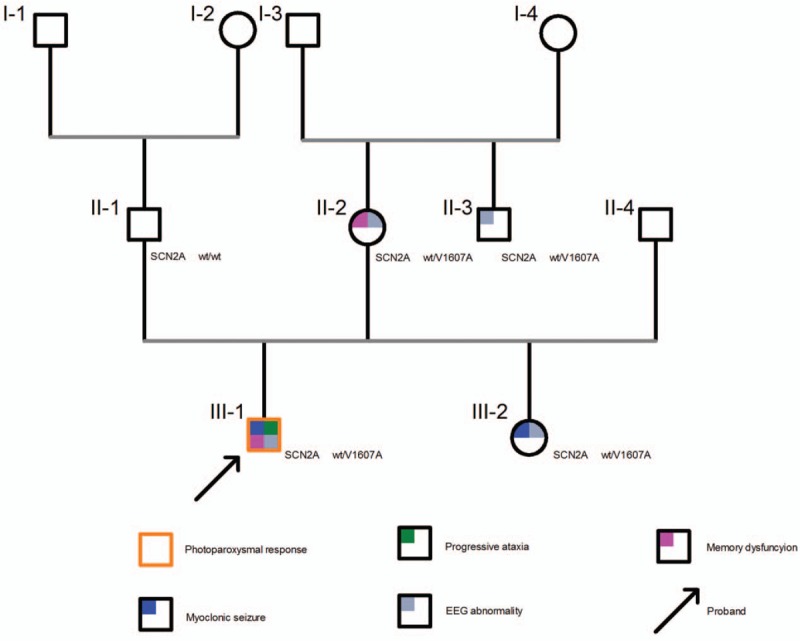
Patient concerns: The 21-year-old male proband suffered from frequent myoclonus at 11 years old with subsequent progressive ataxia. His elder maternal half-sister also experienced myoclonus. Genomic DNA of the patients was extracted from the peripheral blood cells of the proband, elder maternal half-sister, parents, and uncle of the proband. Targeted next-generation sequencing was used to screen gene mutations in the proband. The potential functional effects of mutations within SCN2A were predicted In silico analyses.
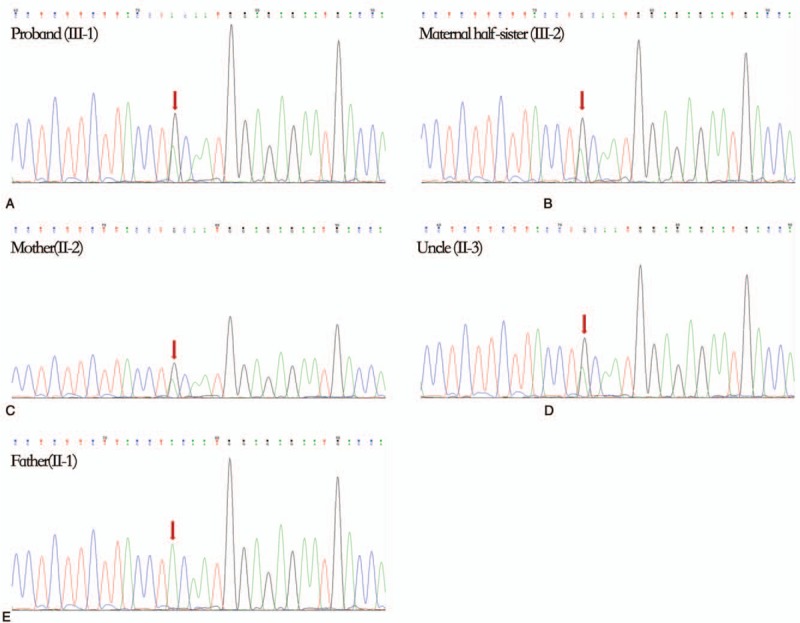
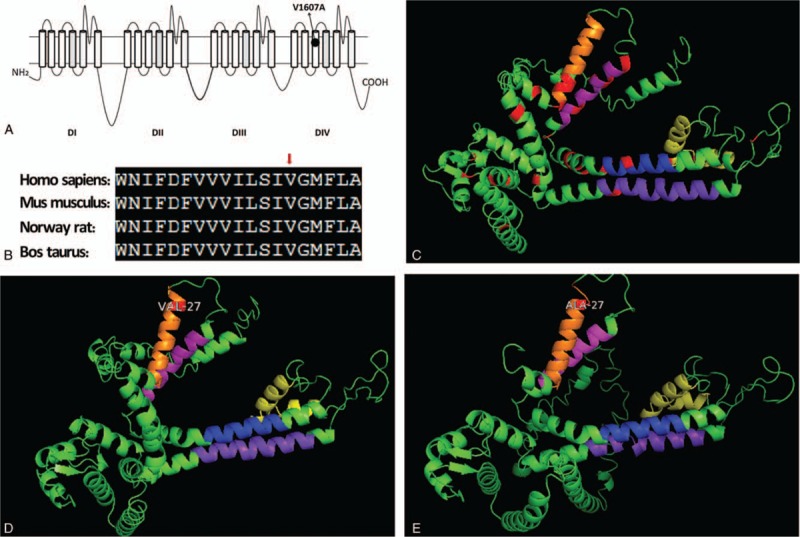
Diagnoses: Genetic testing revealed a novel SCN2A variant, c.T4820C, which contains a highly conserved amino acid substitution within segment S5 (p.V1607A). This mutation was predicted to produce a dysfunctional Nav1.2 protein by Mutation Taster and Protein Variation Effect Analyzer (PROVEAN). Genotype-phenotype correlation showed an incomplete penetrance of p.V1607A.
Interventions: The proband was treated by multiple antiepileptic drugs. These included carbamazepine, oxcarbazepine, valproate, and topiramate.
Outcomes: The duration of follow up was 2 years, and the proband developed drug-resistant epilepsy.
Lessons: The case gives us the lesson that SCN2A mutation can contribute to juvenile-onset myoclonus. Our findings extend the spectrums of SCN2A mutations and the clinical features of patients with SCN2A mutations.
Conflict of interest statement
The authors have no conflicts of interest to disclose.
Figures
Similar articles
-
The phenotype and treatment of SCN2A-related developmental and epileptic encephalopathy.Epileptic Disord. 2020 Oct 1;22(5):563-570. doi: 10.1684/epd.2020.1199. Epileptic Disord. 2020. PMID: 33000761
-
[Phenotype study of SCN2A gene related epilepsy].Zhonghua Er Ke Za Zhi. 2018 Jul 2;56(7):518-523. doi: 10.3760/cma.j.issn.0578-1310.2018.07.009. Zhonghua Er Ke Za Zhi. 2018. PMID: 29996185 Chinese.
-
SCN2A mutation in an infant presenting with migrating focal seizures and infantile spasm responsive to a ketogenic diet.Brain Dev. 2018 Sep;40(8):724-727. doi: 10.1016/j.braindev.2018.03.005. Epub 2018 Apr 4. Brain Dev. 2018. PMID: 29625812
-
The phenotypic spectrum of SCN2A-related epilepsy.Eur J Paediatr Neurol. 2020 Jan;24:117-122. doi: 10.1016/j.ejpn.2019.12.016. Epub 2019 Dec 12. Eur J Paediatr Neurol. 2020. PMID: 31924505 Review.
-
Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities.Epilepsia. 2014 Apr;55(4):e25-9. doi: 10.1111/epi.12554. Epub 2014 Mar 1. Epilepsia. 2014. PMID: 24579881 Review.
Cited by
-
Bromodomain-containing protein 2 gene polymorphism among patients with photosensitive epilepsy in Indonesia.Epilepsia Open. 2025 Apr;10(2):571-580. doi: 10.1002/epi4.70019. Epub 2025 Mar 7. Epilepsia Open. 2025. PMID: 40053300 Free PMC article.
-
Epilepsy and brain channelopathies from infancy to adulthood.Neurol Sci. 2020 Apr;41(4):749-761. doi: 10.1007/s10072-019-04190-x. Epub 2019 Dec 14. Neurol Sci. 2020. PMID: 31838630 Review.
-
Human Cortical Organoids with a Novel SCN2A Variant Exhibit Hyperexcitability and Differential Responses to Anti-Seizure Compounds.Neurosci Bull. 2025 Jun 20. doi: 10.1007/s12264-025-01429-w. Online ahead of print. Neurosci Bull. 2025. PMID: 40540154
References
-
- Wolff M, Johannesen KM, Hedrich UBS, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017;140:1316–36. - PubMed
-
- Horvath GA, Demos M, Shyr C, et al. Secondary neurotransmitter deficiencies in epilepsy caused by voltage-gated sodium channelopathies: a potential treatment target. Mol Genet Metab 2016;117:42–8. - PubMed
-
- Kobayashi K, Ohzono H, Shinohara M, et al. Acute encephalopathy with a novel point mutation in the SCN2A gene. Epilepsy Res 2012;102:109–12. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
