

Covalent Inhibition in Drug Discovery
- PMID: 30816012
- PMCID: PMC6816337
- DOI: 10.1002/cmdc.201900107
Covalent Inhibition in Drug Discovery
Abstract
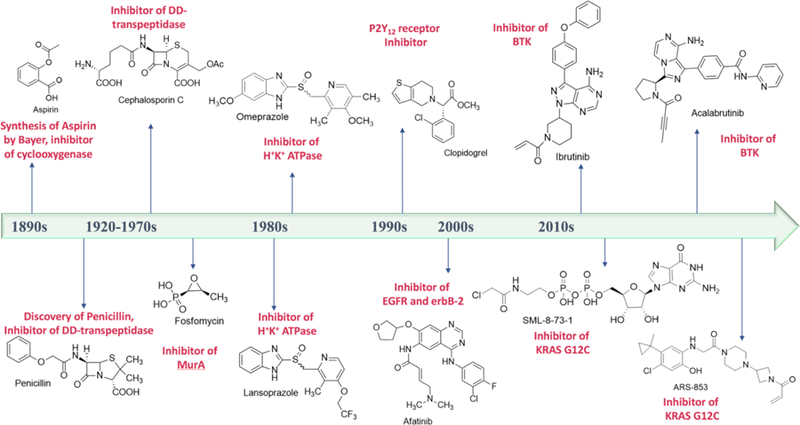
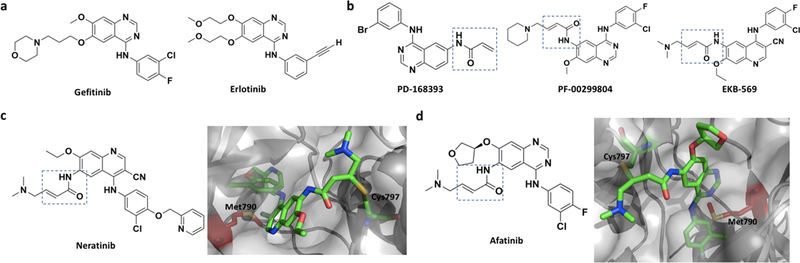
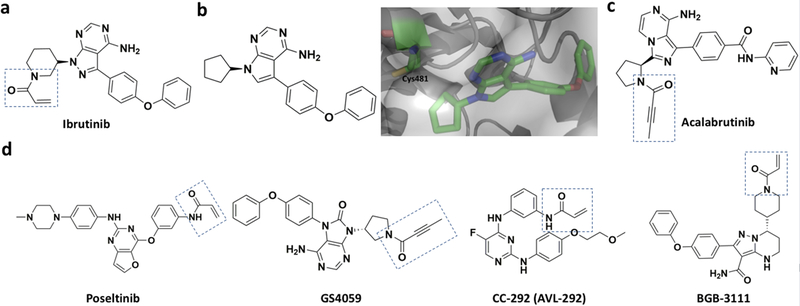
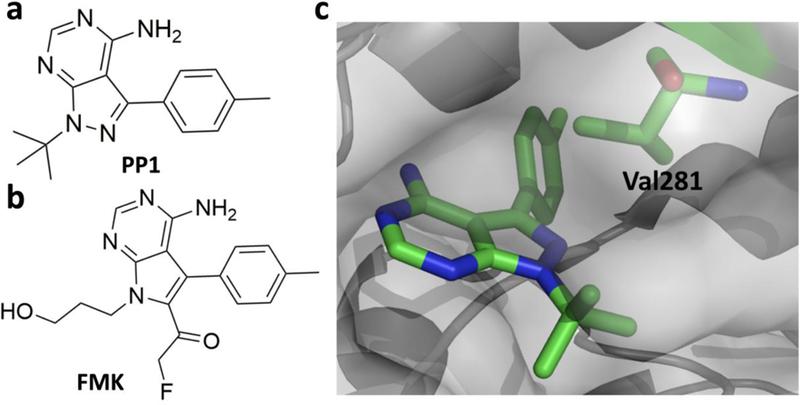
Although covalent inhibitors have been used as therapeutics for more than a century, there has been general resistance in the pharmaceutical industry against their further development due to safety concerns. This inclination has recently been reverted after the development of a wide variety of covalent inhibitors to address human health conditions along with the US Food and Drug Administration (FDA) approval of several covalent therapeutics for use in humans. Along with this exciting resurrection of an old drug discovery concept, this review surveys enzymes that can be targeted by covalent inhibitors for the treatment of human diseases. We focus on protein kinases, RAS proteins, and a few other enzymes that have been studied extensively as targets for covalent inhibition, with the aim to address challenges in designing effective covalent drugs and to provide suggestions in the area that have yet to be explored.
Keywords: RAS proteins; acetylcholinesterase; caspase; cathepsin; covalent inhibitors; protein kinases.
© 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figures



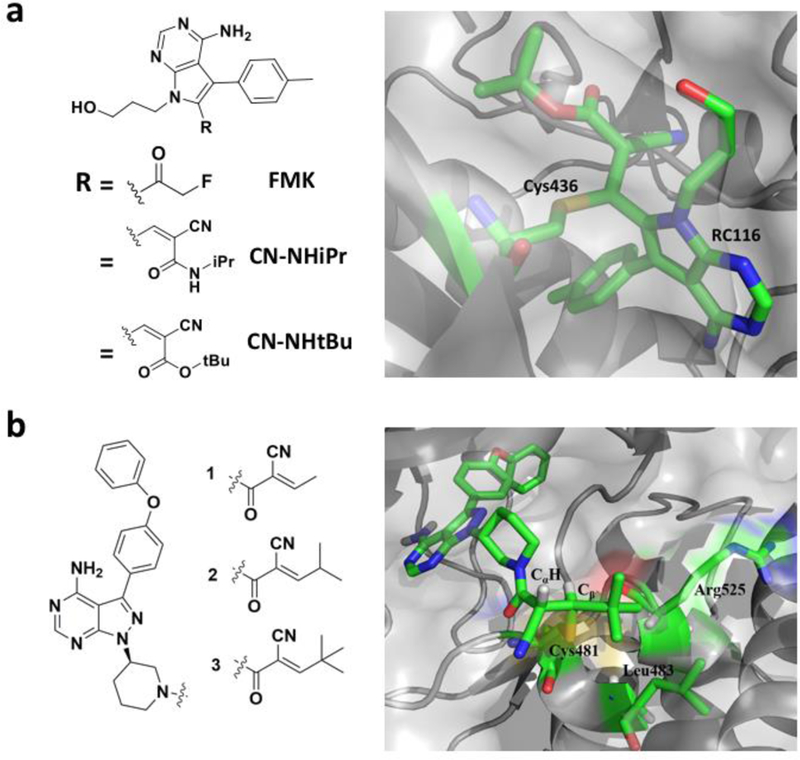
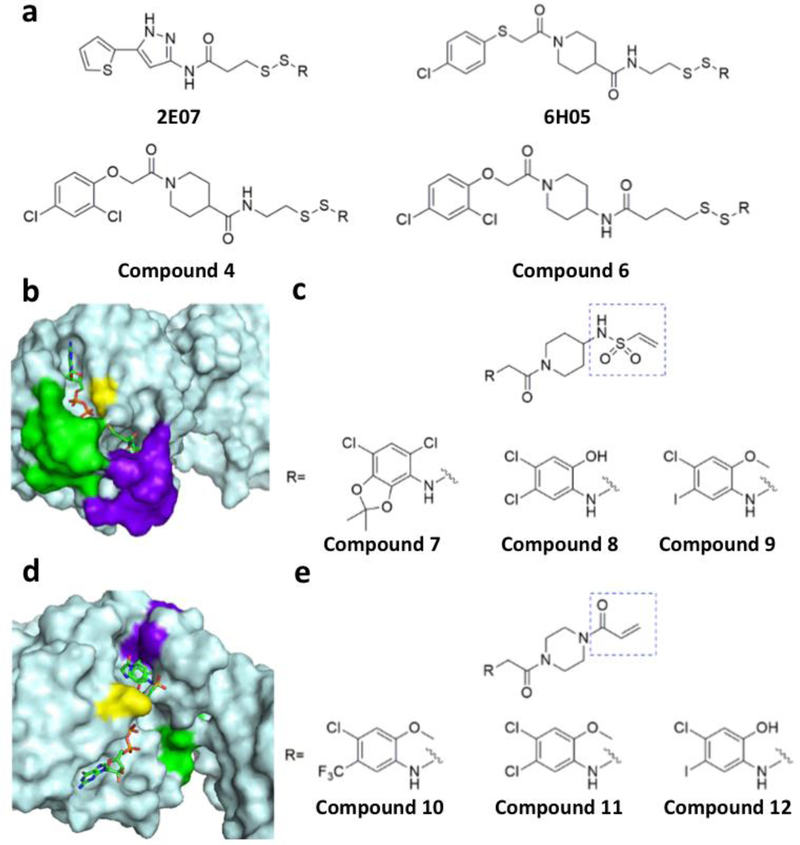
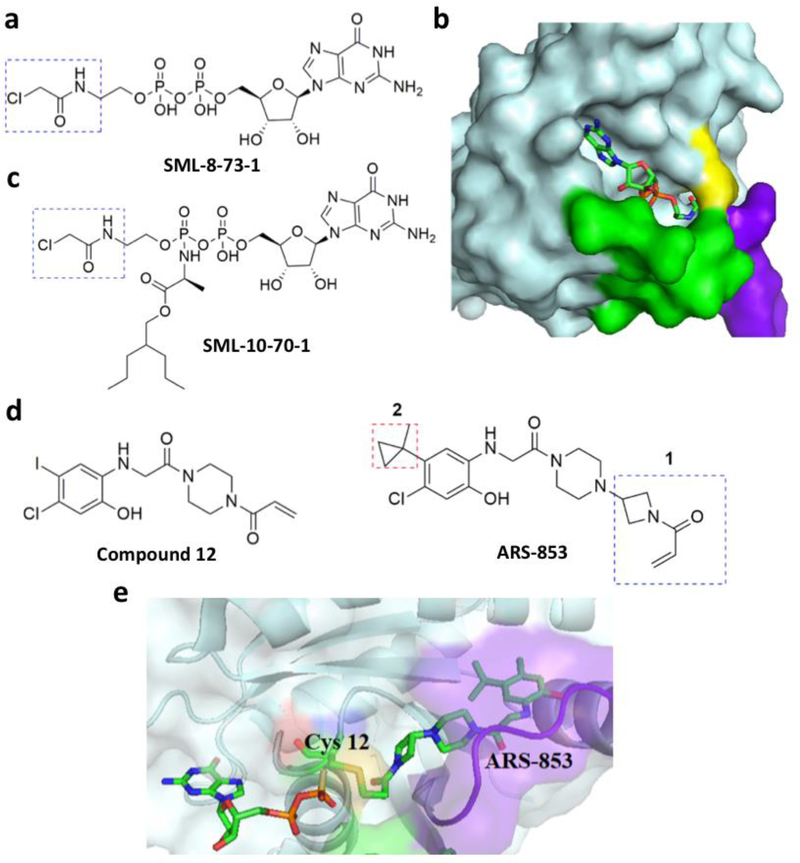
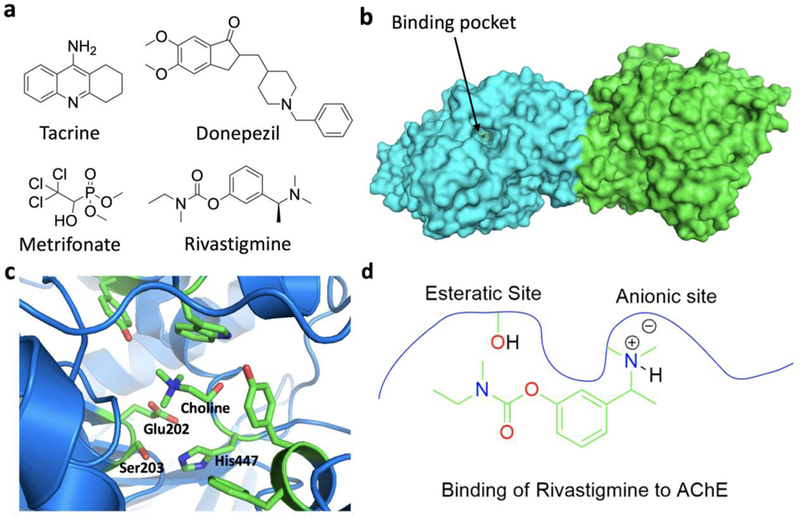

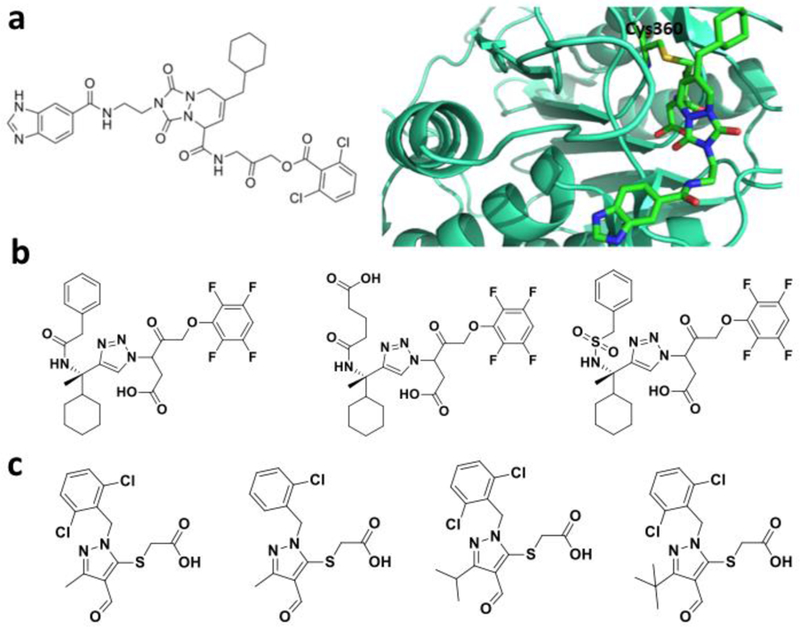
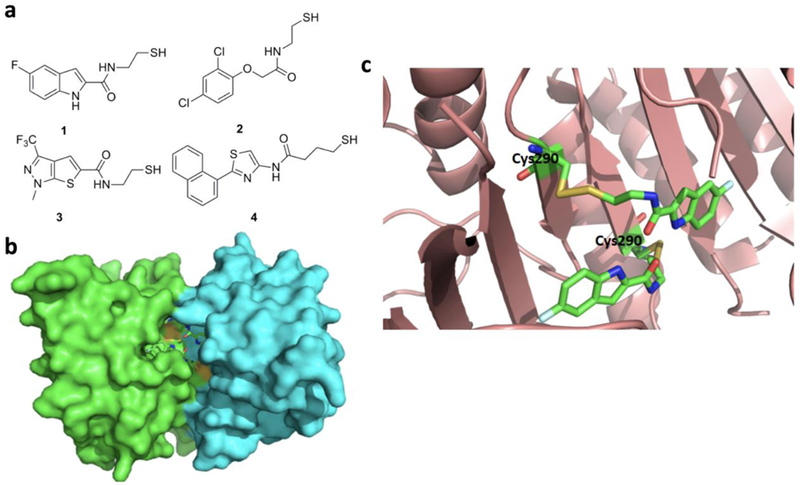
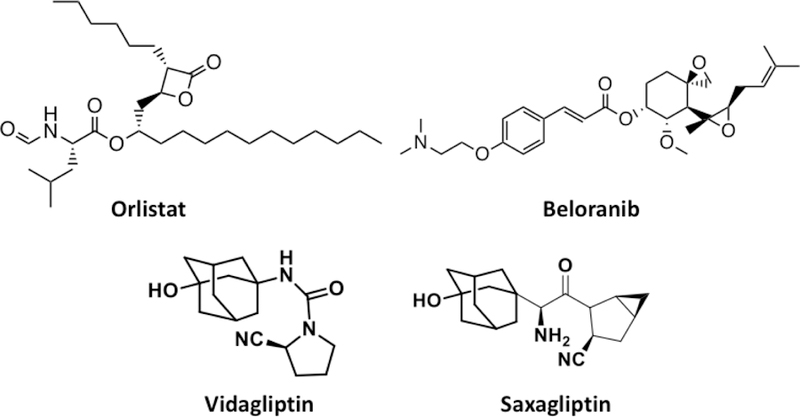
References
-
- Baillie TA, Angew Chem Int Ed Engl 2016, 55, 13408–13421. - PubMed
-
- Singh J, Petter RC, Baillie TA, Whitty A, Nat Rev Drug Discov 2011, 10, 307–317. - PubMed
-
- Picot D, Loll PJ, Garavito RM, Nature 1994, 367, 243–249; P. J. Loll, D. Picot, R. M. Garavito, Nat Struct Biol 1995, 2, 637–643. - PubMed
-
- Baillie TA, Drug Metab Rev 2015, 47, 4–11. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
