

The CDR1 and Other Regions of Immunoglobulin Light Chains are Hot Spots for Amyloid Aggregation
- PMID: 30816248
- PMCID: PMC6395779
- DOI: 10.1038/s41598-019-39781-3
The CDR1 and Other Regions of Immunoglobulin Light Chains are Hot Spots for Amyloid Aggregation
Abstract
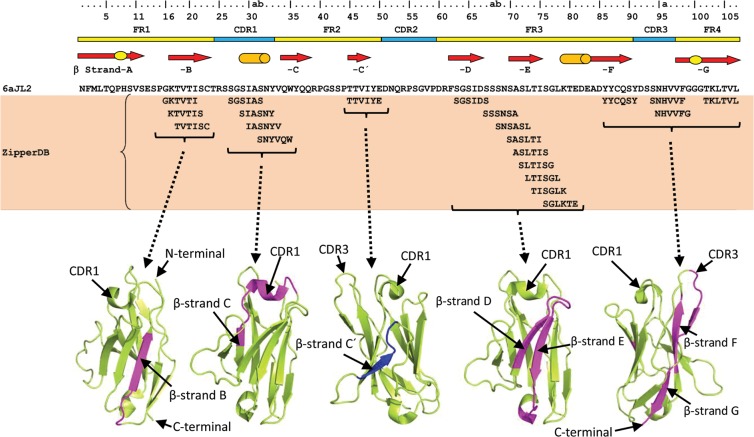
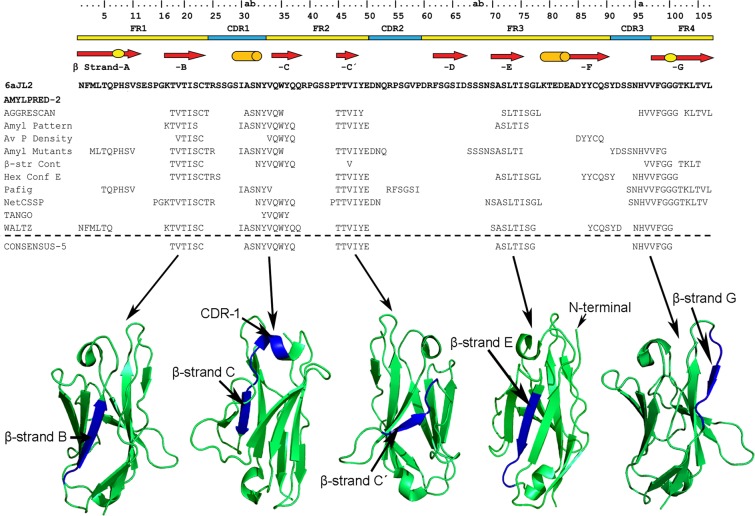
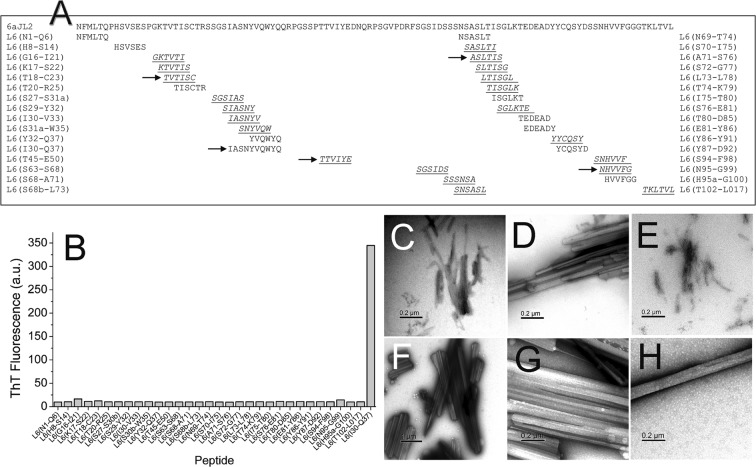
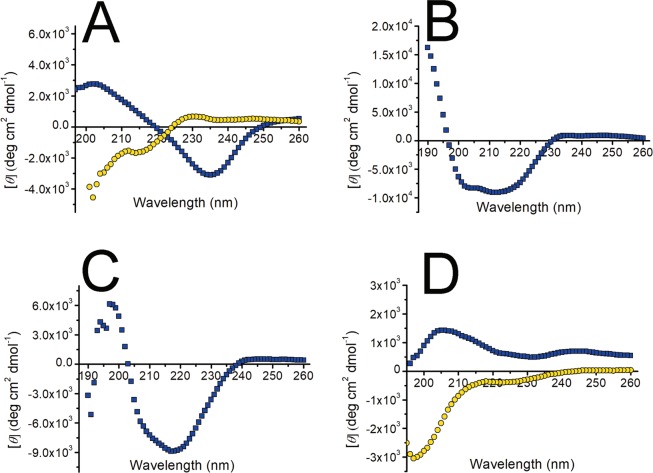
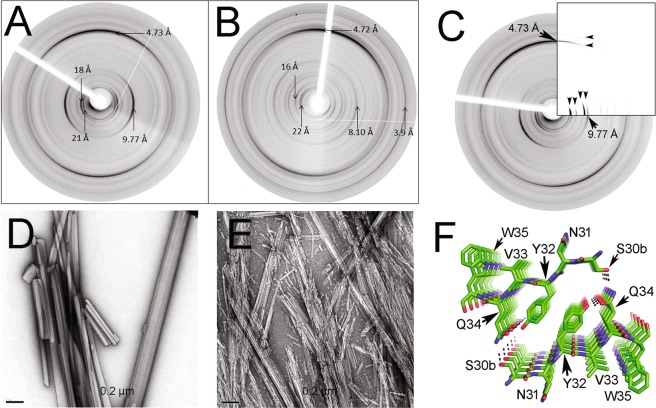
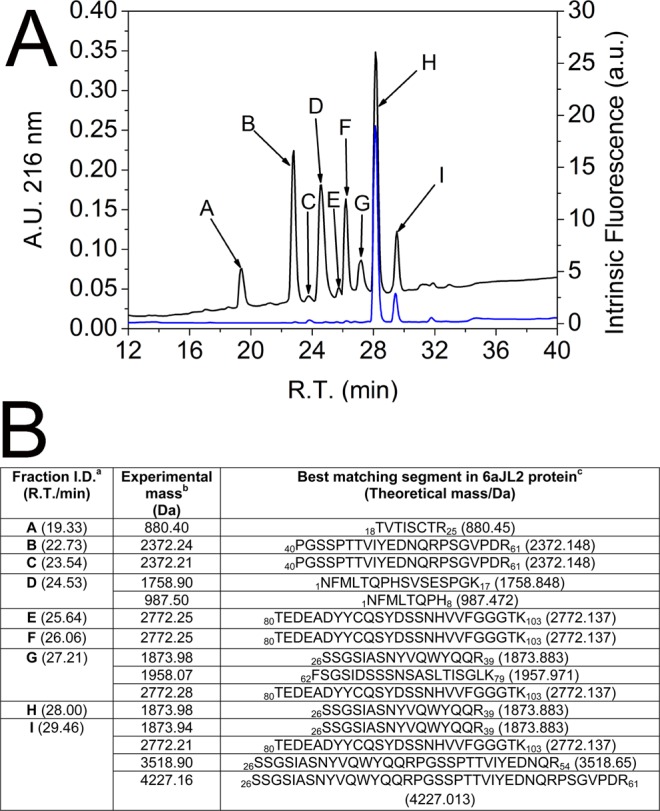
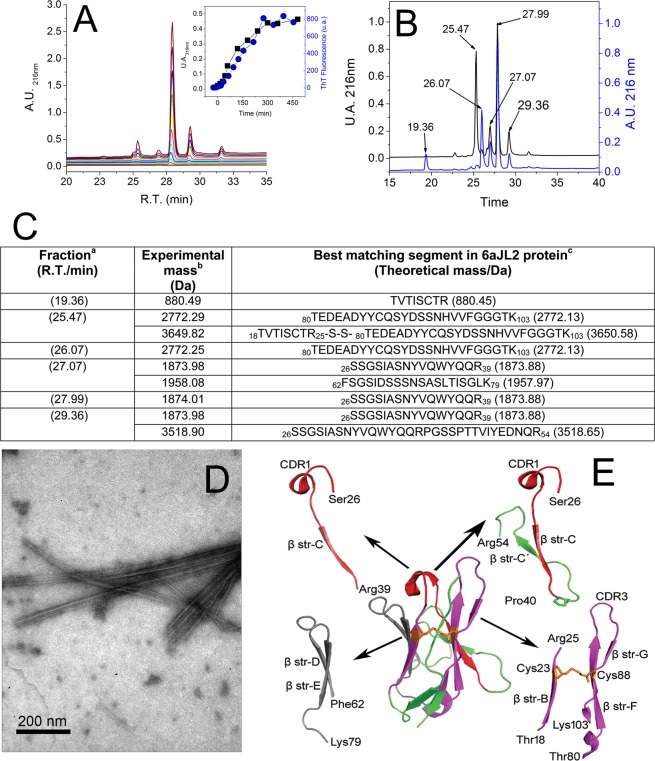
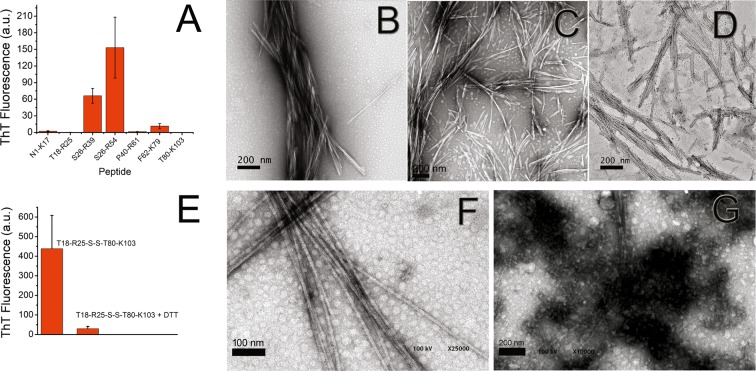
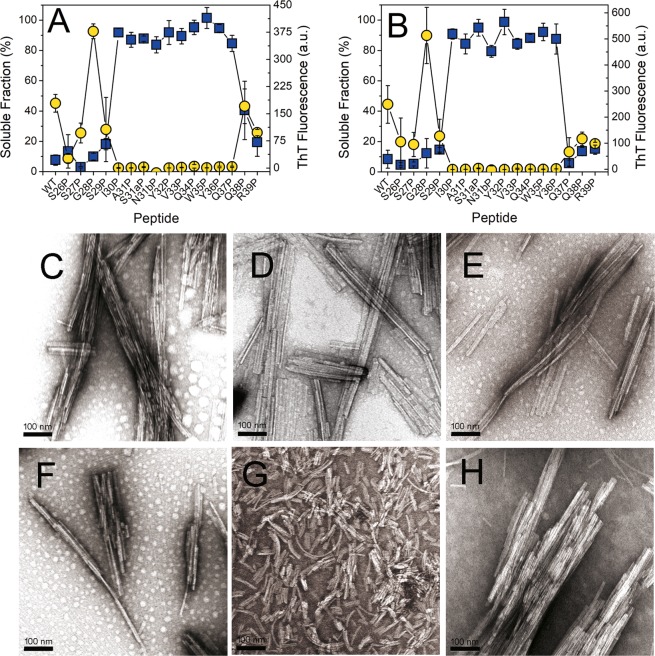
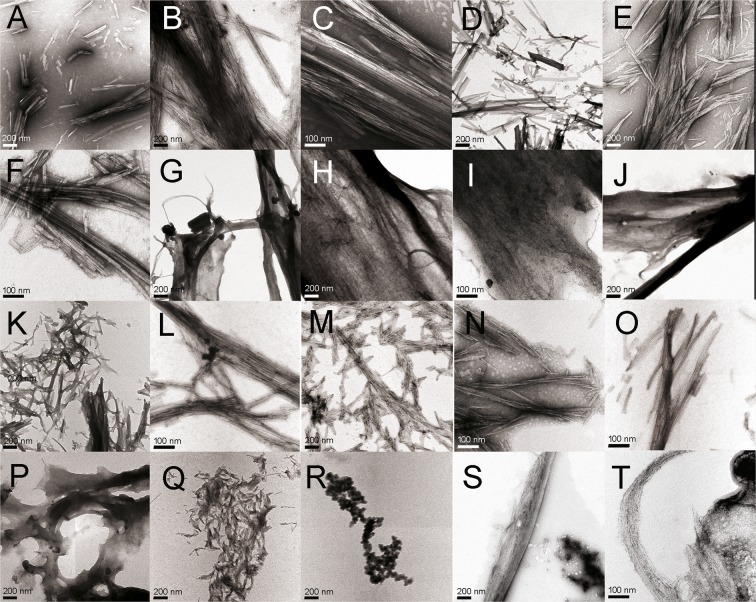
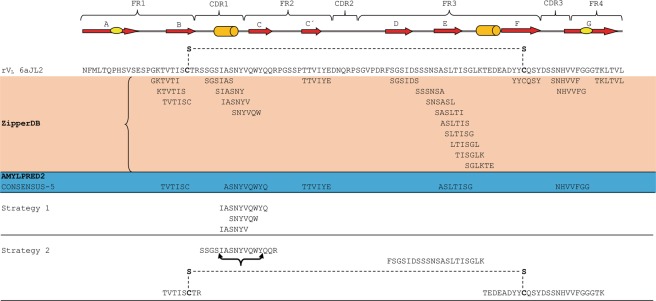
Immunoglobulin light chain-derived (AL) amyloidosis is a debilitating disease without known cure. Almost nothing is known about the structural factors driving the amyloidogenesis of the light chains. This study aimed to identify the fibrillogenic hotspots of the model protein 6aJL2 and in pursuing this goal, two complementary approaches were applied. One of them was based on several web-based computational tools optimized to predict fibrillogenic/aggregation-prone sequences based on different structural and biophysical properties of the polypeptide chain. Then, the predictions were confirmed with an ad-hoc synthetic peptide library. In the second approach, 6aJL2 protein was proteolyzed with trypsin, and the products incubated in aggregation-promoting conditions. Then, the aggregation-prone fragments were identified by combining standard proteomic methods, and the results validated with a set of synthetic peptides with the sequence of the tryptic fragments. Both strategies coincided to identify a fibrillogenic hotspot located at the CDR1 and β-strand C of the protein, which was confirmed by scanning proline mutagenesis analysis. However, only the proteolysis-based strategy revealed additional fibrillogenic hotspots in two other regions of the protein. It was shown that a fibrillogenic hotspot associated to the CDR1 is also encoded by several κ and λ germline variable domain gene segments. Some parts of this study have been included in the chapter "The Structural Determinants of the Immunoglobulin Light Chain Amyloid Aggregation", published in Physical Biology of Proteins and Peptides, Springer 2015 (ISBN 978-3-319-21687-4).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Identification of two principal amyloid-driving segments in variable domains of Ig light chains in systemic light-chain amyloidosis.J Biol Chem. 2018 Dec 21;293(51):19659-19671. doi: 10.1074/jbc.RA118.004142. Epub 2018 Oct 24. J Biol Chem. 2018. PMID: 30355736 Free PMC article.
-
Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient.Nat Commun. 2019 Mar 20;10(1):1269. doi: 10.1038/s41467-019-09133-w. Nat Commun. 2019. PMID: 30894521 Free PMC article.
-
Mass spectrometry characterization of light chain fragmentation sites in cardiac AL amyloidosis: insights into the timing of proteolysis.J Biol Chem. 2020 Dec 4;295(49):16572-16584. doi: 10.1074/jbc.RA120.013461. Epub 2020 Sep 20. J Biol Chem. 2020. PMID: 32952127 Free PMC article.
-
Immunoglobulin light chain amyloid aggregation.Chem Commun (Camb). 2018 Sep 20;54(76):10664-10674. doi: 10.1039/c8cc04396e. Chem Commun (Camb). 2018. PMID: 30087961 Free PMC article. Review.
-
The Process of Amyloid Formation due to Monoclonal Immunoglobulins.Hematol Oncol Clin North Am. 2020 Dec;34(6):1041-1054. doi: 10.1016/j.hoc.2020.07.003. Epub 2020 Sep 12. Hematol Oncol Clin North Am. 2020. PMID: 33099422 Review.
Cited by
-
Dynamic protein structures in normal function and pathologic misfolding in systemic amyloidosis.Biophys Chem. 2022 Jan;280:106699. doi: 10.1016/j.bpc.2021.106699. Epub 2021 Oct 14. Biophys Chem. 2022. PMID: 34773861 Free PMC article. Review.
-
Role of the mechanisms for antibody repertoire diversification in monoclonal light chain deposition disorders: when a friend becomes foe.Front Immunol. 2023 Jul 13;14:1203425. doi: 10.3389/fimmu.2023.1203425. eCollection 2023. Front Immunol. 2023. PMID: 37520549 Free PMC article. Review.
-
Molecular mechanism of amyloidogenic mutations in hypervariable regions of antibody light chains.J Biol Chem. 2021 Jan-Jun;296:100334. doi: 10.1016/j.jbc.2021.100334. Epub 2021 Jan 26. J Biol Chem. 2021. PMID: 33508322 Free PMC article.
-
Seeded fibrils of the germline variant of human λ-III immunoglobulin light chain FOR005 have a similar core as patient fibrils with reduced stability.J Biol Chem. 2020 Dec 25;295(52):18474-18484. doi: 10.1074/jbc.RA120.016006. Epub 2020 Oct 22. J Biol Chem. 2020. PMID: 33093170 Free PMC article.
-
Construction of a Large Size Human Immunoglobulin Heavy Chain Variable (VH) Domain Library, Isolation and Characterization of Novel Human Antibody VH Domains Targeting PD-L1 and CD22.Front Immunol. 2022 Apr 6;13:869825. doi: 10.3389/fimmu.2022.869825. eCollection 2022. Front Immunol. 2022. PMID: 35464476 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
