

De novo and inherited TCF20 pathogenic variants are associated with intellectual disability, dysmorphic features, hypotonia, and neurological impairments with similarities to Smith-Magenis syndrome
- PMID: 30819258
- PMCID: PMC6393995
- DOI: 10.1186/s13073-019-0623-0
De novo and inherited TCF20 pathogenic variants are associated with intellectual disability, dysmorphic features, hypotonia, and neurological impairments with similarities to Smith-Magenis syndrome
Erratum in
-
Correction to: De novo and inherited TCF20 pathogenic variants are associated with intellectual disability, dysmorphic features, hypotonia, and neurological impairments with similarities to Smith-Magenis syndrome.Genome Med. 2019 Mar 25;11(1):16. doi: 10.1186/s13073-019-0630-1. Genome Med. 2019. PMID: 30909959 Free PMC article.
Abstract
Background: Neurodevelopmental disorders are genetically and phenotypically heterogeneous encompassing developmental delay (DD), intellectual disability (ID), autism spectrum disorders (ASDs), structural brain abnormalities, and neurological manifestations with variants in a large number of genes (hundreds) associated. To date, a few de novo mutations potentially disrupting TCF20 function in patients with ID, ASD, and hypotonia have been reported. TCF20 encodes a transcriptional co-regulator structurally related to RAI1, the dosage-sensitive gene responsible for Smith-Magenis syndrome (deletion/haploinsufficiency) and Potocki-Lupski syndrome (duplication/triplosensitivity).
Methods: Genome-wide analyses by exome sequencing (ES) and chromosomal microarray analysis (CMA) identified individuals with heterozygous, likely damaging, loss-of-function alleles in TCF20. We implemented further molecular and clinical analyses to determine the inheritance of the pathogenic variant alleles and studied the spectrum of phenotypes.
Results: We report 25 unique inactivating single nucleotide variants/indels (1 missense, 1 canonical splice-site variant, 18 frameshift, and 5 nonsense) and 4 deletions of TCF20. The pathogenic variants were detected in 32 patients and 4 affected parents from 31 unrelated families. Among cases with available parental samples, the variants were de novo in 20 instances and inherited from 4 symptomatic parents in 5, including in one set of monozygotic twins. Two pathogenic loss-of-function variants were recurrent in unrelated families. Patients presented with a phenotype characterized by developmental delay, intellectual disability, hypotonia, variable dysmorphic features, movement disorders, and sleep disturbances.
Conclusions: TCF20 pathogenic variants are associated with a novel syndrome manifesting clinical characteristics similar to those observed in Smith-Magenis syndrome. Together with previously described cases, the clinical entity of TCF20-associated neurodevelopmental disorders (TAND) emerges from a genotype-driven perspective.
Keywords: 22q13; Deletions; Haploinsufficiency; Loss-of-function variants; Neurodevelopmental disorders; Smith–Magenis syndrome; TCF20.
Conflict of interest statement
Ethics approval and consent to participate
All participants provided written informed consent to participate in the study. The study was approved by the Institutional Review Board of Baylor College of Medicine (H-22769 and H-41191) and the UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The research conforms with the principles of the Declaration of Helsinki.
Consent for publication
The consent to publish all identifiable information presented in the study including Fig. 2 was provided by the parents or legal guardians of the subjects.
Competing interests
Baylor College of Medicine (BCM) and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), which performs chromosomal microarray analysis and clinical exome sequencing. JAR, SHE, WB, FX, YY, CME and PL are employees of BCM and derive support through a professional services agreement with BG. FV and WZ are employees of BG. JRL serves on the Scientific Advisory Board of BG. JRL has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, and is a coinventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. The other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
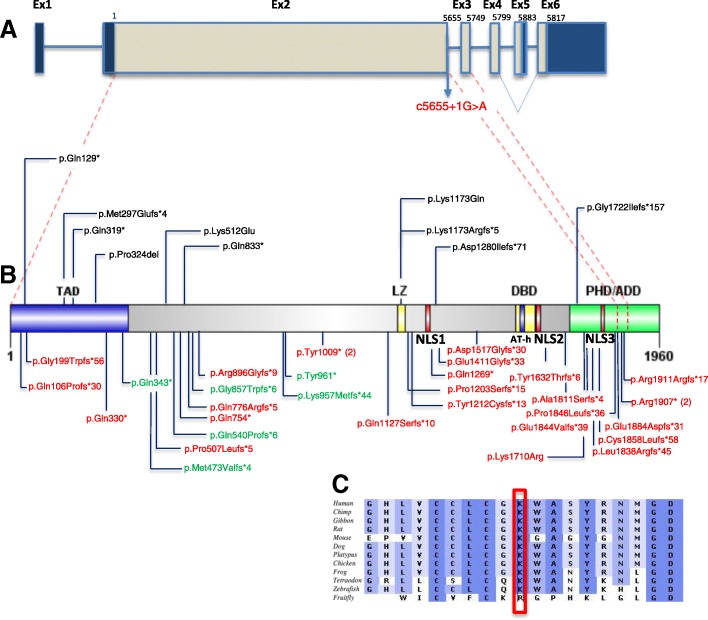


Comment in
-
Molecular basis for phenotypic similarity of genetic disorders.Genome Med. 2019 Apr 23;11(1):24. doi: 10.1186/s13073-019-0641-y. Genome Med. 2019. PMID: 31014384 Free PMC article.
References
-
- Simenson K, Oiglane-Shlik E, Teek R, Kuuse K, Ounap KA, et al. A patient with the classic features of Phelan-McDermid syndrome and a high immunoglobulin E level caused by a cryptic interstitial 0.72-Mb deletion in the 22q13.2 region. Am J Med Genet A. 2014;164A(3):806–809. doi: 10.1002/ajmg.a.36358. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
