

Atherogenic LOX-1 signaling is controlled by SPPL2-mediated intramembrane proteolysis
- PMID: 30819724
- PMCID: PMC6446863
- DOI: 10.1084/jem.20171438
Atherogenic LOX-1 signaling is controlled by SPPL2-mediated intramembrane proteolysis
Abstract
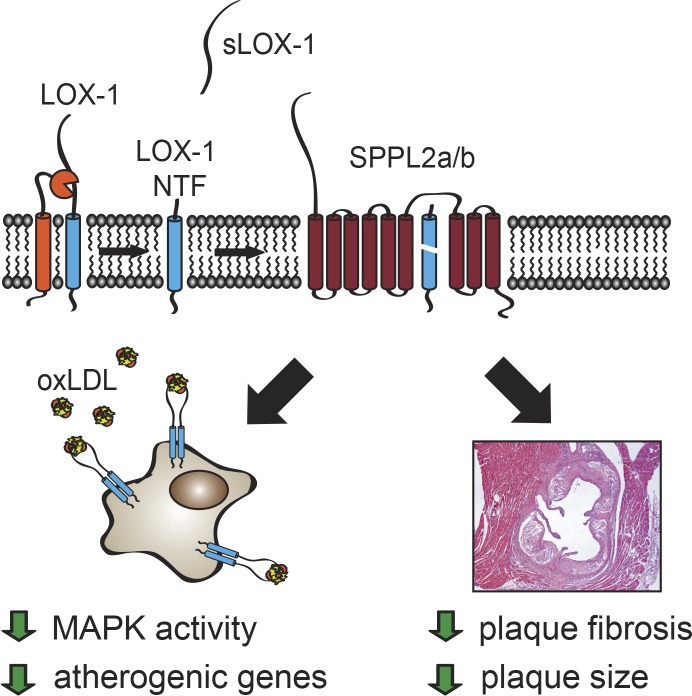
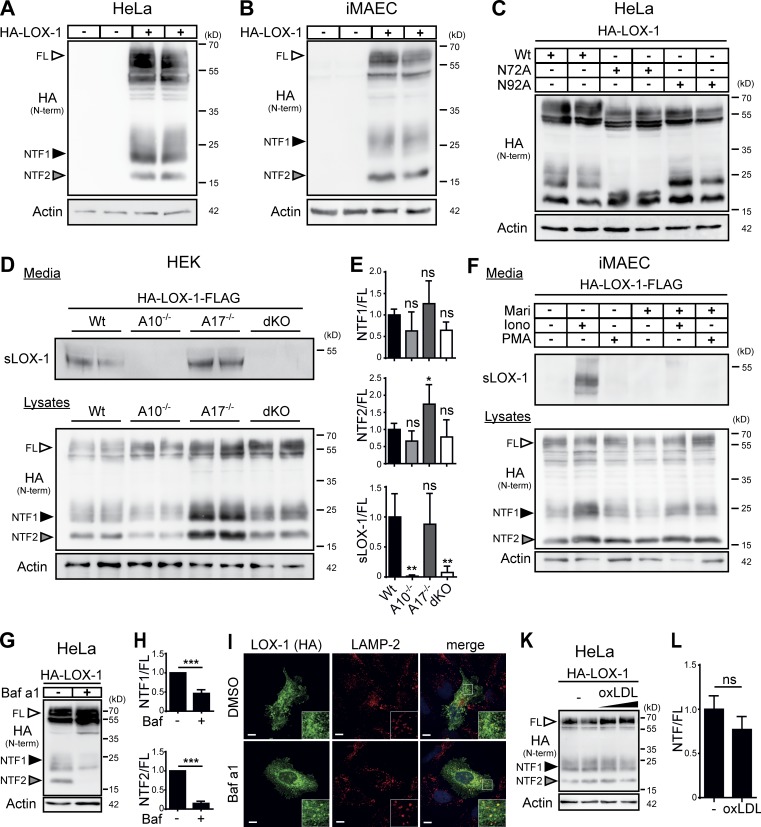
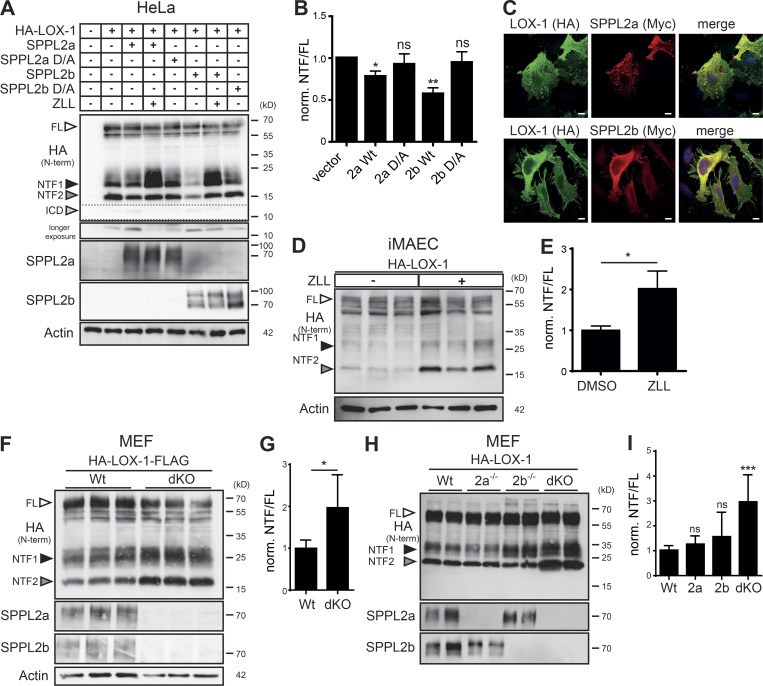
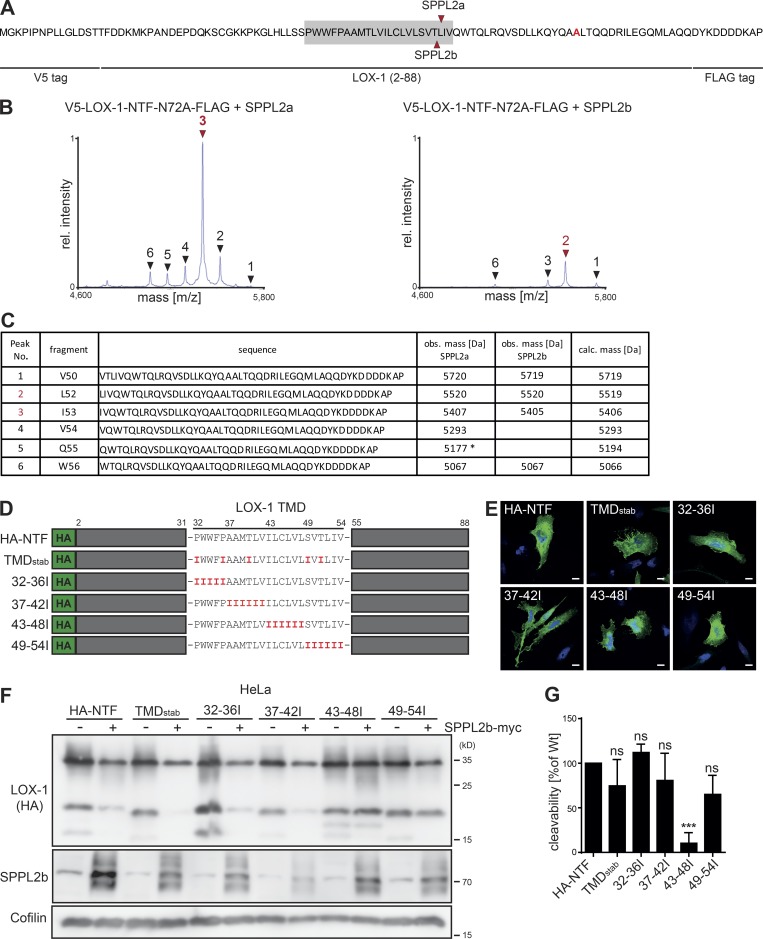
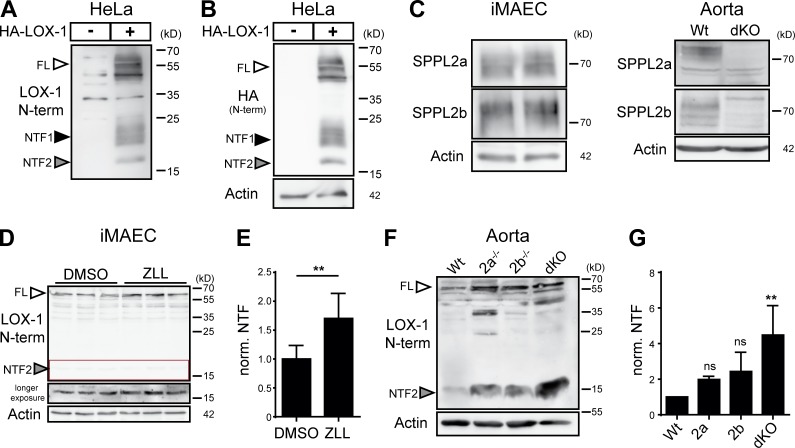
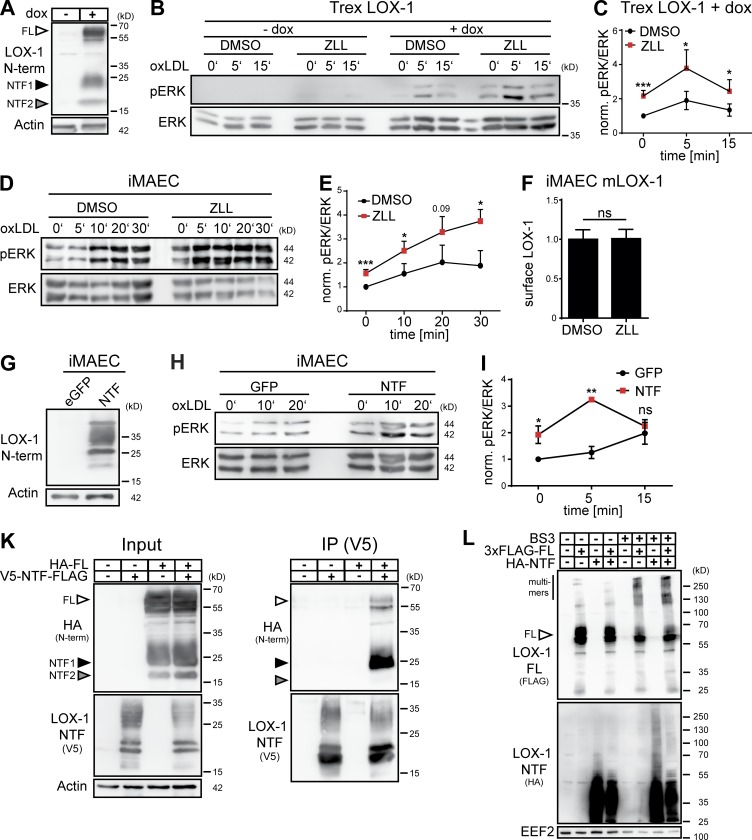
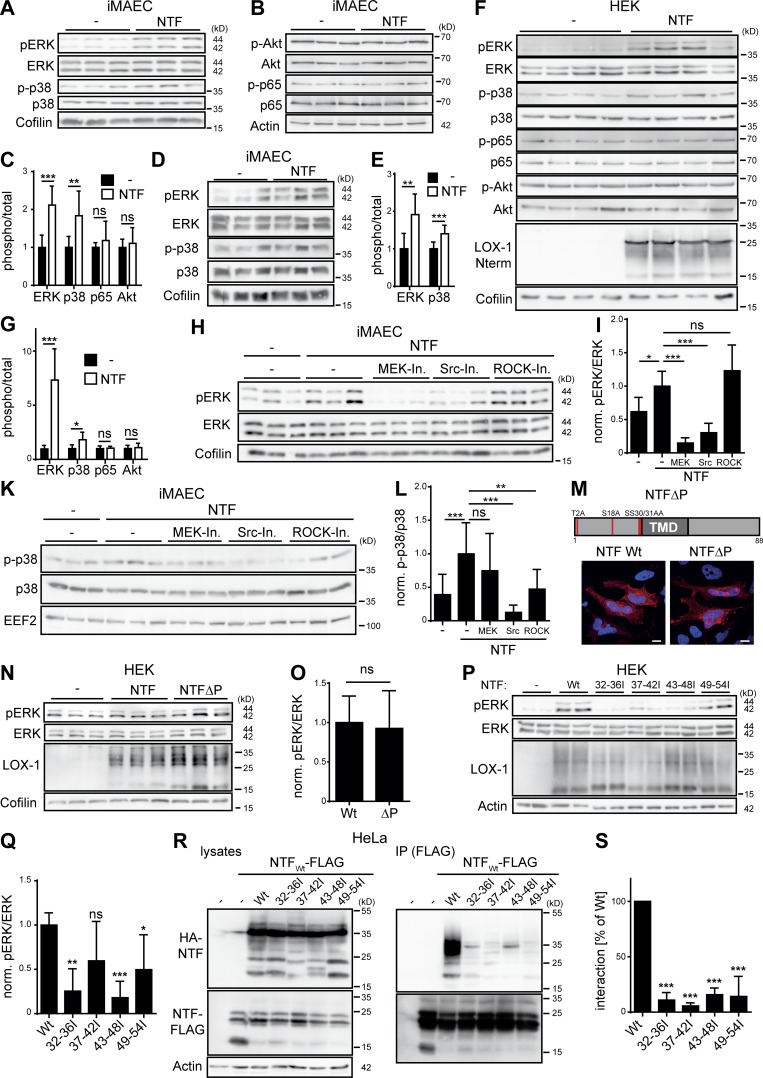
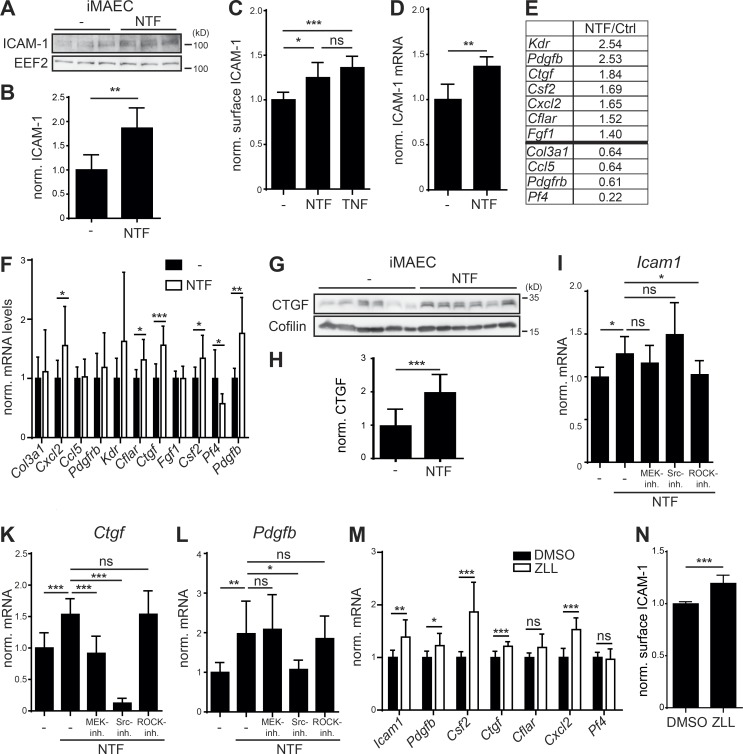
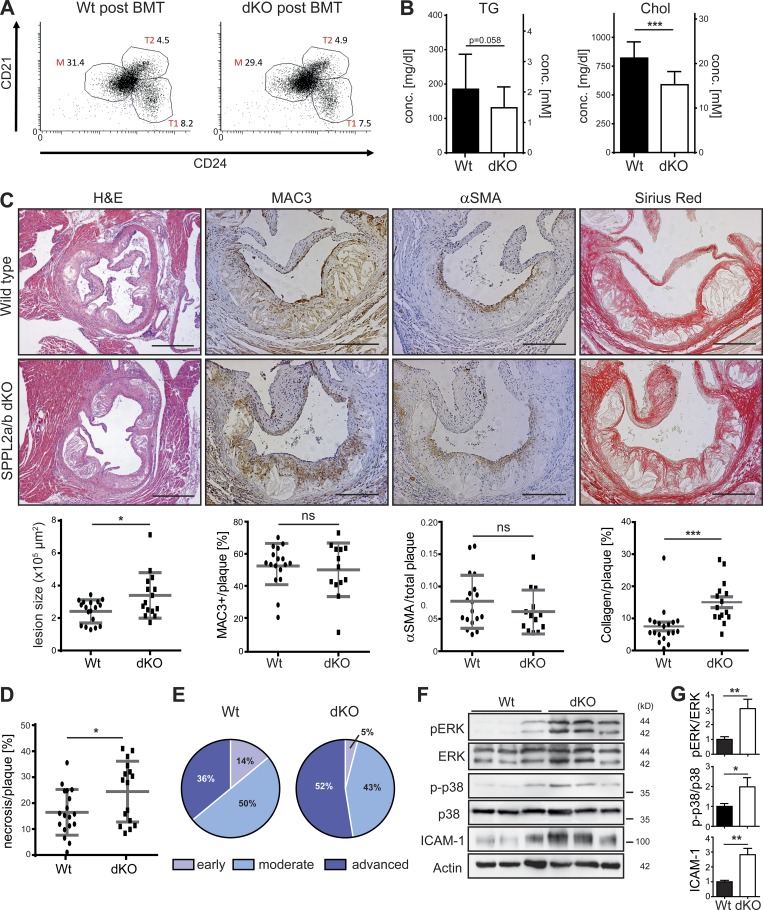
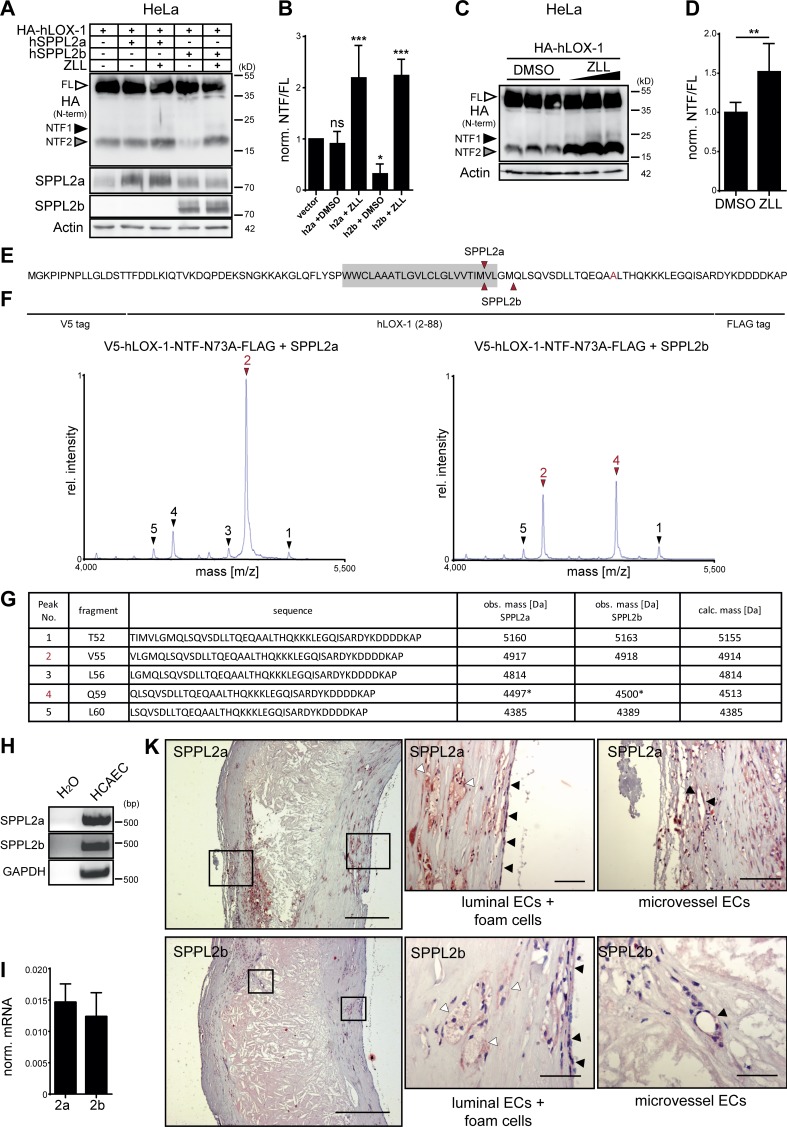
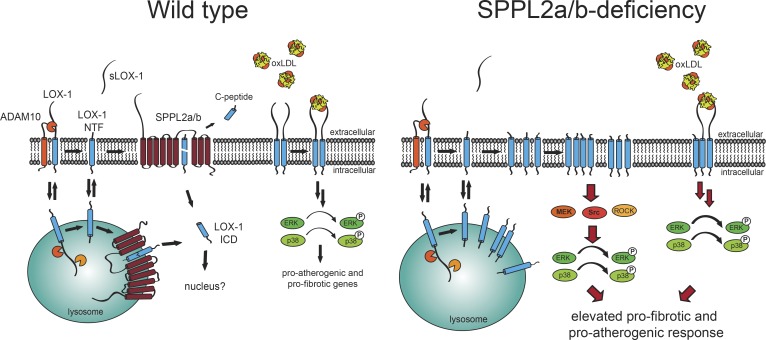
The lectin-like oxidized LDL receptor 1 (LOX-1) is a key player in the development of atherosclerosis. LOX-1 promotes endothelial activation and dysfunction by mediating uptake of oxidized LDL and inducing pro-atherogenic signaling. However, little is known about modulators of LOX-1-mediated responses. Here, we show that the function of LOX-1 is controlled proteolytically. Ectodomain shedding by the metalloprotease ADAM10 and lysosomal degradation generate membrane-bound N-terminal fragments (NTFs), which we identified as novel substrates of the intramembrane proteases signal peptide peptidase-like 2a and b (SPPL2a/b). SPPL2a/b control cellular LOX-1 NTF levels which, following self-association via their transmembrane domain, can activate MAP kinases in a ligand-independent manner. This leads to an up-regulation of several pro-atherogenic and pro-fibrotic targets including ICAM-1 and the connective tissue growth factor CTGF. Consequently, SPPL2a/b-deficient mice, which accumulate LOX-1 NTFs, develop larger and more advanced atherosclerotic plaques than controls. This identifies intramembrane proteolysis by SPPL2a/b as a novel atheroprotective mechanism via negative regulation of LOX-1 signaling.
© 2019 Mentrup et al.
Figures
References
-
- Akhmedov A., Rozenberg I., Paneni F., Camici G.G., Shi Y., Doerries C., Sledzinska A., Mocharla P., Breitenstein A., Lohmann C., et al. . 2014. Endothelial overexpression of LOX-1 increases plaque formation and promotes atherosclerosis in vivo. Eur. Heart J. 35:2839–2848. 10.1093/eurheartj/eht532 - DOI - PubMed
-
- Al-Alwan L.A., Chang Y., Mogas A., Halayko A.J., Baglole C.J., Martin J.G., Rousseau S., Eidelman D.H., and Hamid Q.. 2013. Differential roles of CXCL2 and CXCL3 and their receptors in regulating normal and asthmatic airway smooth muscle cell migration. J. Immunol. 191:2731–2741. 10.4049/jimmunol.1203421 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
