

Synergy from gene expression and network mining (SynGeNet) method predicts synergistic drug combinations for diverse melanoma genomic subtypes
- PMID: 30820351
- PMCID: PMC6391384
- DOI: 10.1038/s41540-019-0085-4
Synergy from gene expression and network mining (SynGeNet) method predicts synergistic drug combinations for diverse melanoma genomic subtypes
Abstract
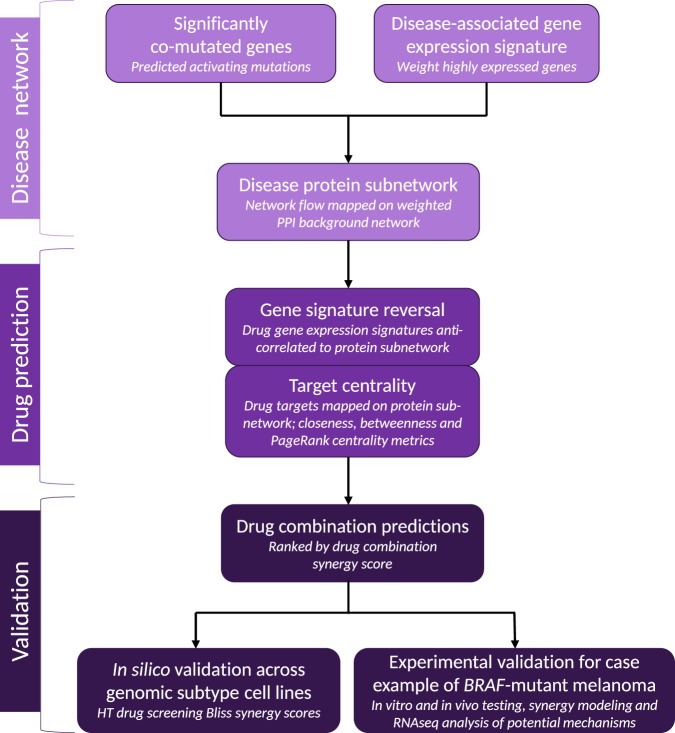
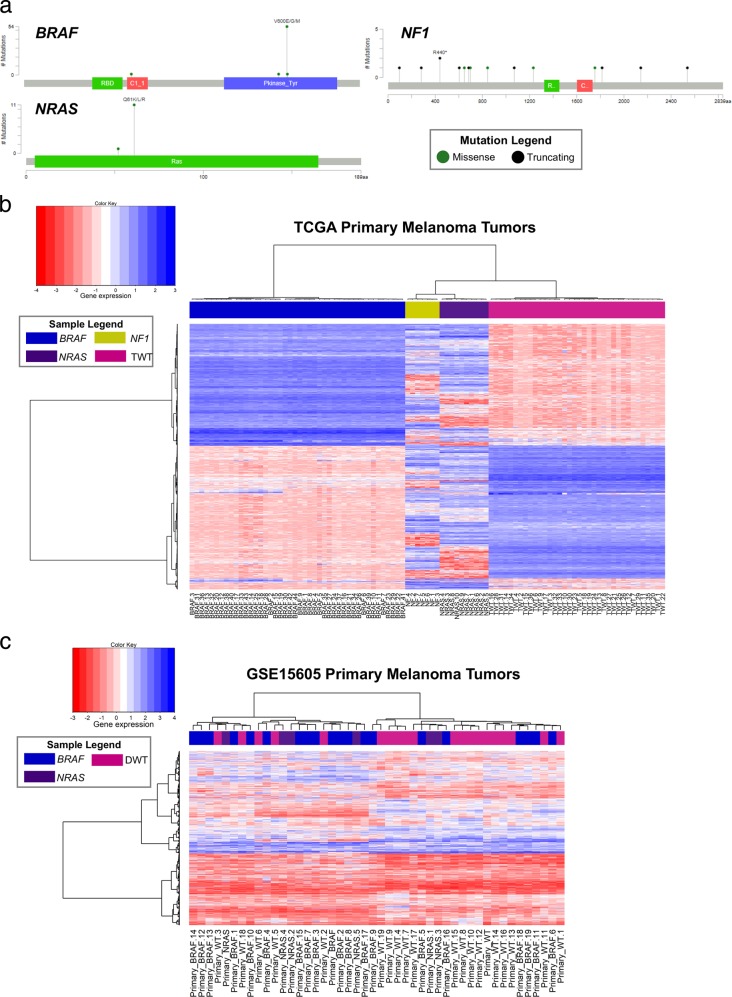
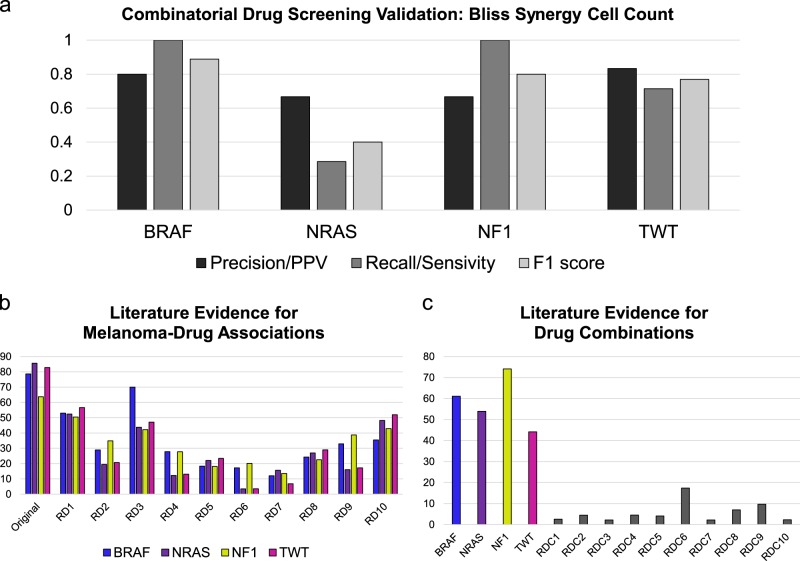
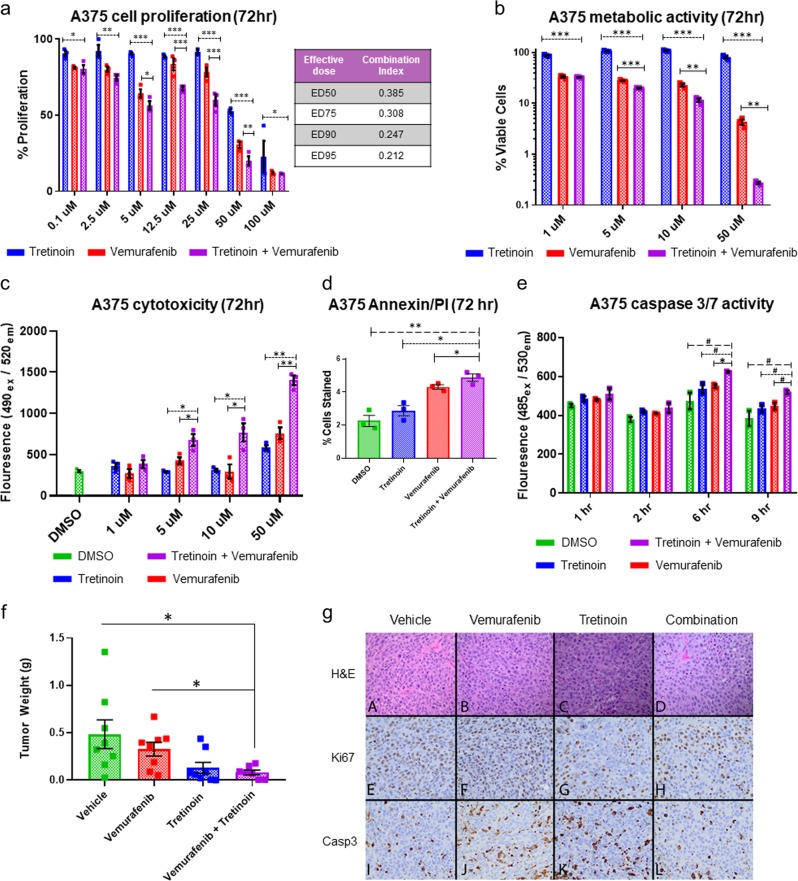
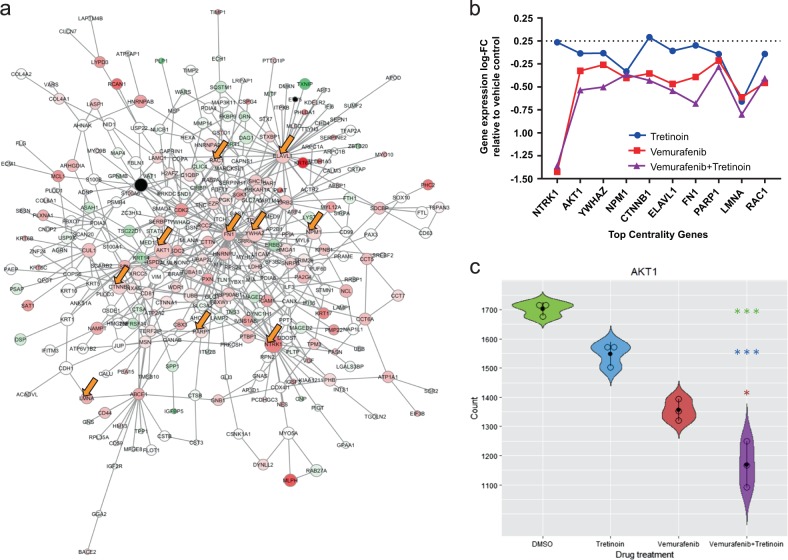
Systems biology perspectives are crucial for understanding the pathophysiology of complex diseases, and therefore hold great promise for the discovery of novel treatment strategies. Drug combinations have been shown to improve durability and reduce resistance to available first-line therapies in a variety of cancers; however, traditional drug discovery approaches are prohibitively cost and labor-intensive to evaluate large-scale matrices of potential drug combinations. Computational methods are needed to efficiently model complex interactions of drug target pathways and identify mechanisms underlying drug combination synergy. In this study, we employ a computational approach, SynGeNet (Synergy from Gene expression and Network mining), which integrates transcriptomics-based connectivity mapping and network centrality analysis to analyze disease networks and predict drug combinations. As an exemplar of a disease in which combination therapies demonstrate efficacy in genomic-specific contexts, we investigate malignant melanoma. We employed SynGeNet to generate drug combination predictions for each of the four major genomic subtypes of melanoma (BRAF, NRAS, NF1, and triple wild type) using publicly available gene expression and mutation data. We validated synergistic drug combinations predicted by our method across all genomic subtypes using results from a high-throughput drug screening study across. Finally, we prospectively validated the drug combination for BRAF-mutant melanoma that was top ranked by our approach, vemurafenib (BRAF inhibitor) + tretinoin (retinoic acid receptor agonist), using both in vitro and in vivo models of BRAF-mutant melanoma and RNA-sequencing analysis of drug-treated melanoma cells to validate the predicted mechanisms. Our approach is applicable to a wide range of disease domains, and, importantly, can model disease-relevant protein subnetworks in precision medicine contexts.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
