

Journey to the center of the protein: allostery from multitemperature multiconformer X-ray crystallography
- PMID: 30821702
- PMCID: PMC6400254
- DOI: 10.1107/S2059798318017941
Journey to the center of the protein: allostery from multitemperature multiconformer X-ray crystallography
Abstract
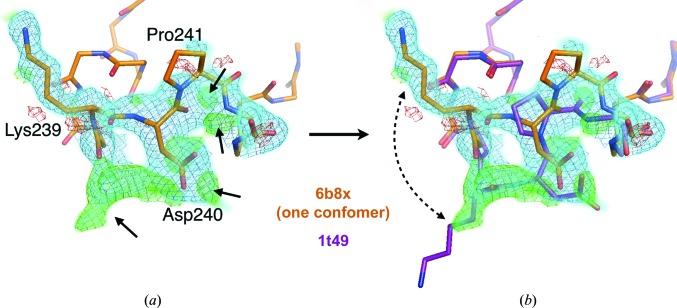
Proteins inherently fluctuate between conformations to perform functions in the cell. For example, they sample product-binding, transition-state-stabilizing and product-release states during catalysis, and they integrate signals from remote regions of the structure for allosteric regulation. However, there is a lack of understanding of how these dynamic processes occur at the basic atomic level. This gap can be at least partially addressed by combining variable-temperature (instead of traditional cryogenic temperature) X-ray crystallography with algorithms for modeling alternative conformations based on electron-density maps, in an approach called multitemperature multiconformer X-ray crystallography (MMX). Here, the use of MMX to reveal alternative conformations at different sites in a protein structure and to estimate the degree of energetic coupling between them is discussed. These insights can suggest testable hypotheses about allosteric mechanisms. Temperature is an easily manipulated experimental parameter, so the MMX approach is widely applicable to any protein that yields well diffracting crystals. Moreover, the general principles of MMX are extensible to other perturbations such as pH, pressure, ligand concentration etc. Future work will explore strategies for leveraging X-ray data across such perturbation series to more quantitatively measure how different parts of a protein structure are coupled to each other, and the consequences thereof for allostery and other aspects of protein function.
Keywords: allostery; conformational heterogeneity; multiconformer modeling; multitemperature crystallography; protein flexibility.
open access.
Figures





References
-
- Babcock, N. S., Keedy, D. A., Fraser, J. S. & Sivak, D. A. (2018). bioRxiv, 448795.
-
- Barends, T. R. M., Foucar, L., Ardevol, A., Nass, K., Aquila, A., Botha, S., Doak, R. B., Falahati, K., Hartmann, E., Hilpert, M., Heinz, M., Hoffmann, M. C., Köfinger, J., Koglin, J. E., Kovacsova, G., Liang, M., Milathianaki, D., Lemke, H. T., Reinstein, J., Roome, C. M., Shoeman, R. L., Williams, G. J., Burghardt, I., Hummer, G., Boutet, S. & Schlichting, I. (2015). Science, 350, 445–450. - PubMed
-
- Baxter, E. L., Aguila, L., Alonso-Mori, R., Barnes, C. O., Bonagura, C. A., Brehmer, W., Brunger, A. T., Calero, G., Caradoc-Davies, T. T., Chatterjee, R., Degrado, W. F., Fraser, J. S., Ibrahim, M., Kern, J., Kobilka, B. K., Kruse, A. C., Larsson, K. M., Lemke, H. T., Lyubimov, A. Y., Manglik, A., McPhillips, S. E., Norgren, E., Pang, S. S., Soltis, S. M., Song, J., Thomaston, J., Tsai, Y., Weis, W. I., Woldeyes, R. A., Yachandra, V., Yano, J., Zouni, A. & Cohen, A. E. (2016). Acta Cryst. D72, 2–11.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
