

MMSplice: modular modeling improves the predictions of genetic variant effects on splicing
- PMID: 30823901
- PMCID: PMC6396468
- DOI: 10.1186/s13059-019-1653-z
MMSplice: modular modeling improves the predictions of genetic variant effects on splicing
Abstract
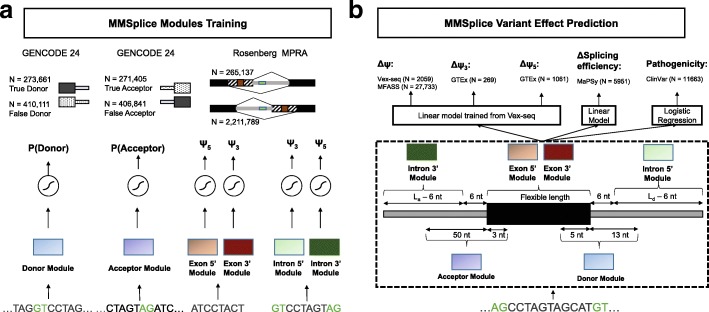
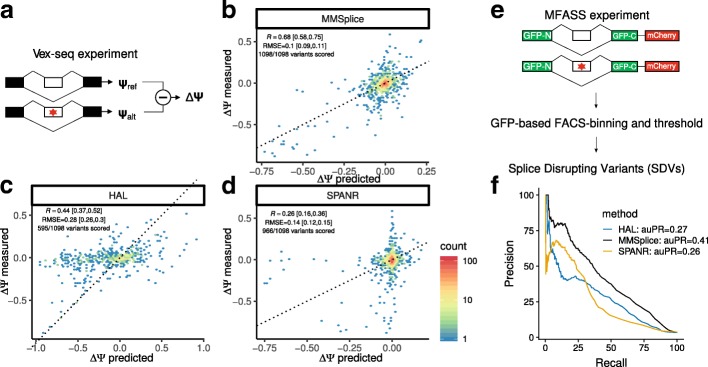
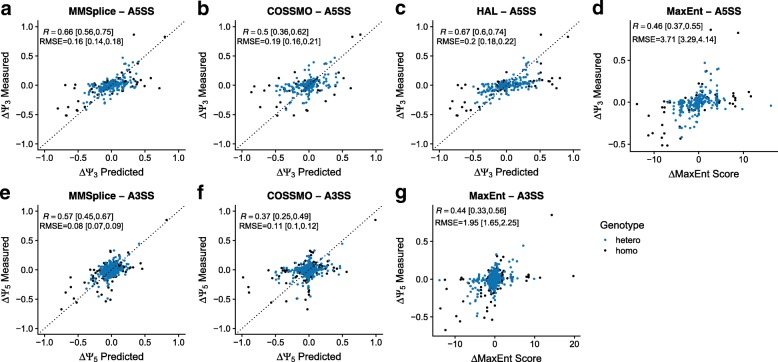
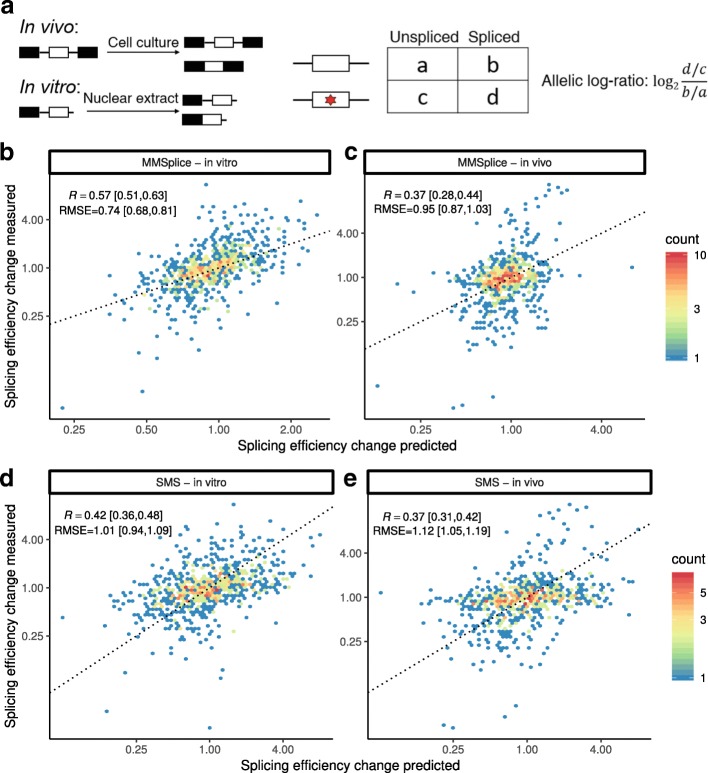
Predicting the effects of genetic variants on splicing is highly relevant for human genetics. We describe the framework MMSplice (modular modeling of splicing) with which we built the winning model of the CAGI5 exon skipping prediction challenge. The MMSplice modules are neural networks scoring exon, intron, and splice sites, trained on distinct large-scale genomics datasets. These modules are combined to predict effects of variants on exon skipping, splice site choice, splicing efficiency, and pathogenicity, with matched or higher performance than state-of-the-art. Our models, available in the repository Kipoi, apply to variants including indels directly from VCF files.
Keywords: Deep learning; Modular modeling; Splicing; Variant effect; Variant pathogenicity.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- López-Bigas N, Audit B, Ouzounis C, Parra G, Guigó R. Are splicing mutations the most frequent cause of hereditary disease?FEBS Lett. 2005; 579(9):1900–3. 10.1016/j.febslet.2005.02.047. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
