

Selecting variants of unknown significance through network-based gene-association significantly improves risk prediction for disease-control cohorts
- PMID: 30824863
- PMCID: PMC6397233
- DOI: 10.1038/s41598-019-39796-w
Selecting variants of unknown significance through network-based gene-association significantly improves risk prediction for disease-control cohorts
Abstract
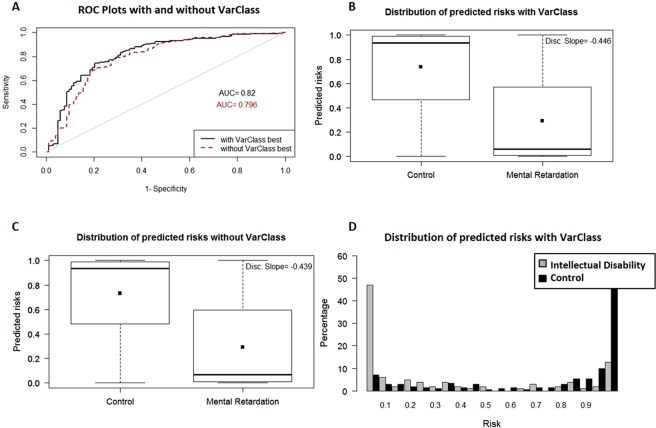
Variants of unknown/uncertain significance (VUS) pose a huge dilemma in current genetic variation screening methods and genetic counselling. Driven by methods of next generation sequencing (NGS) such as whole exome sequencing (WES), a plethora of VUS are being detected in research laboratories as well as in the health sector. Motivated by this overabundance of VUS, we propose a novel computational methodology, termed VariantClassifier (VarClass), which utilizes gene-association networks and polygenic risk prediction models to shed light into this grey area of genetic variation in association with disease. VarClass has been evaluated using numerous validation steps and proves to be very successful in assigning significance to VUS in association with specific diseases of interest. Notably, using VUS that are deemed significant by VarClass, we improved risk prediction accuracy in four large case-studies involving disease-control cohorts from GWAS as well as WES, when compared to traditional odds ratio analysis. Biological interpretation of selected high scoring VUS revealed interesting biological themes relevant to the diseases under investigation. VarClass is available as a standalone tool for large-scale data analyses, as well as a web-server with additional functionalities through a user-friendly graphical interface.
Conflict of interest statement
The authors declare no competing interests.
Figures





Similar articles
-
BRCA1 and BRCA2 Testing through Next Generation Sequencing in a Small Cohort of Italian Breast/Ovarian Cancer Patients: Novel Pathogenic and Unknown Clinical Significance Variants.Int J Mol Sci. 2019 Jul 12;20(14):3442. doi: 10.3390/ijms20143442. Int J Mol Sci. 2019. PMID: 31336956 Free PMC article.
-
Exome Sequencing in a Family with Luminal-Type Breast Cancer Underpinned by Variation in the Methylation Pathway.Int J Mol Sci. 2017 Feb 22;18(2):467. doi: 10.3390/ijms18020467. Int J Mol Sci. 2017. PMID: 28241424 Free PMC article.
-
The challenge for the next generation of medical geneticists.Hum Mutat. 2014 Aug;35(8):909-11. doi: 10.1002/humu.22592. Epub 2014 Jun 28. Hum Mutat. 2014. PMID: 24838402 Review.
-
Reporting practices for variants of uncertain significance from next generation sequencing technologies.Eur J Med Genet. 2017 Oct;60(10):553-558. doi: 10.1016/j.ejmg.2017.07.016. Epub 2017 Aug 1. Eur J Med Genet. 2017. PMID: 28774848
-
Variants of uncertain significance in the era of high-throughput genome sequencing: a lesson from breast and ovary cancers.J Exp Clin Cancer Res. 2020 Mar 4;39(1):46. doi: 10.1186/s13046-020-01554-6. J Exp Clin Cancer Res. 2020. PMID: 32127026 Free PMC article. Review.
Cited by
-
Multicenter Consensus Approach to Evaluation of Neonatal Hypotonia in the Genomic Era: A Review.JAMA Neurol. 2022 Apr 1;79(4):405-413. doi: 10.1001/jamaneurol.2022.0067. JAMA Neurol. 2022. PMID: 35254387 Free PMC article. Review.
-
Targeting mutations in cancer.J Clin Invest. 2022 Apr 15;132(8):e154943. doi: 10.1172/JCI154943. J Clin Invest. 2022. PMID: 35426374 Free PMC article. Review.
-
Basic science methods for the characterization of variants of uncertain significance in hypertrophic cardiomyopathy.Front Cardiovasc Med. 2023 Aug 1;10:1238515. doi: 10.3389/fcvm.2023.1238515. eCollection 2023. Front Cardiovasc Med. 2023. PMID: 37600050 Free PMC article. Review.
-
The Role of Genetics in the Management of Heart Failure Patients.Int J Mol Sci. 2023 Oct 16;24(20):15221. doi: 10.3390/ijms242015221. Int J Mol Sci. 2023. PMID: 37894902 Free PMC article. Review.
-
Whole genome sequencing of low input circulating cell-free DNA obtained from normal human subjects.Physiol Rep. 2021 Aug;9(15):e14993. doi: 10.14814/phy2.14993. Physiol Rep. 2021. PMID: 34350716 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
