

Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics
- PMID: 30827679
- PMCID: PMC6408267
- DOI: 10.1016/j.cell.2019.01.022
Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics
Abstract
Lineage tracing provides key insights into the fate of individual cells in complex organisms. Although effective genetic labeling approaches are available in model systems, in humans, most approaches require detection of nuclear somatic mutations, which have high error rates, limited scale, and do not capture cell state information. Here, we show that somatic mutations in mtDNA can be tracked by single-cell RNA or assay for transposase accessible chromatin (ATAC) sequencing. We leverage somatic mtDNA mutations as natural genetic barcodes and demonstrate their utility as highly accurate clonal markers to infer cellular relationships. We track native human cells both in vitro and in vivo and relate clonal dynamics to gene expression and chromatin accessibility. Our approach should allow clonal tracking at a 1,000-fold greater scale than with nuclear genome sequencing, with simultaneous information on cell state, opening the way to chart cellular dynamics in human health and disease.
Keywords: chronic myeloid leukemia; colon cancer; hematopoiesis; lineage tracing; mitochondrial DNA; mtDNA; sequencing; single cell genomics; somatic mutations.
Copyright © 2019 Elsevier Inc. All rights reserved.
Figures
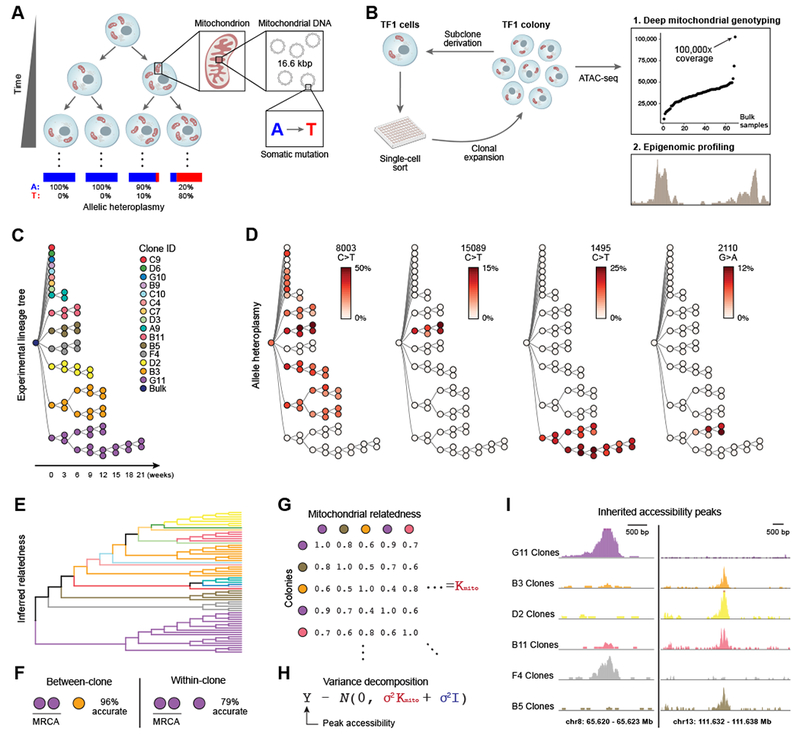

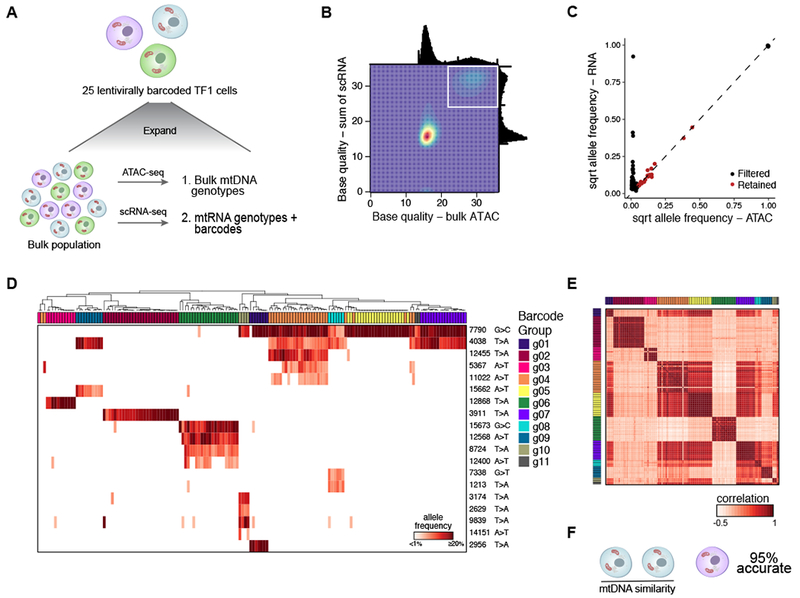
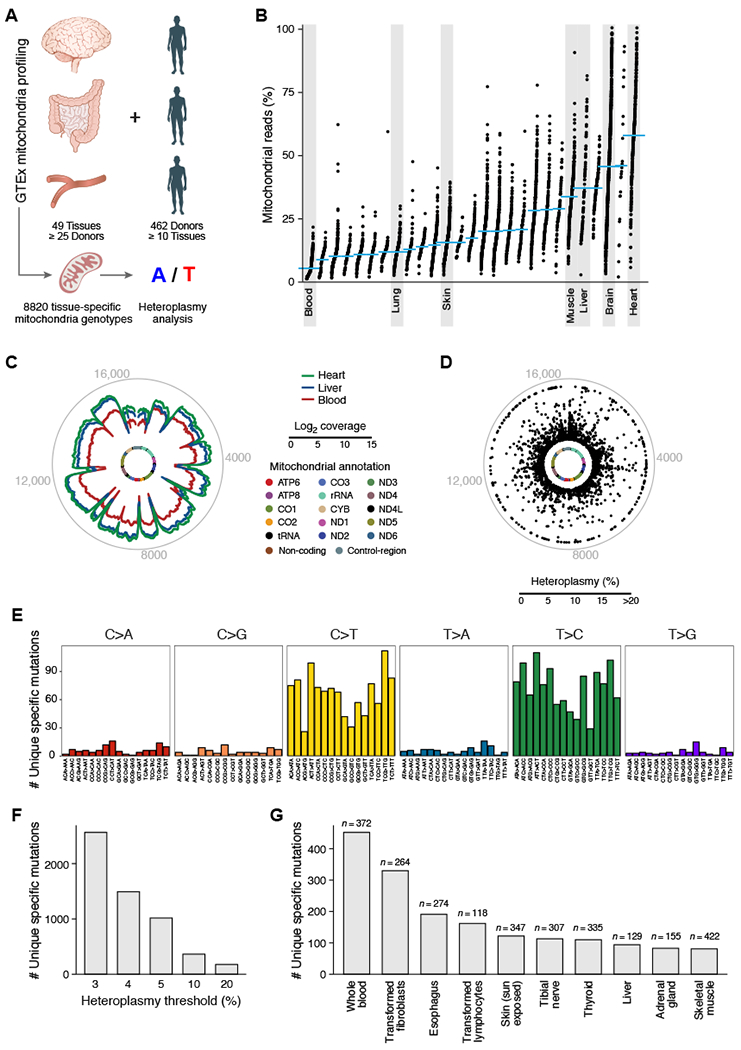
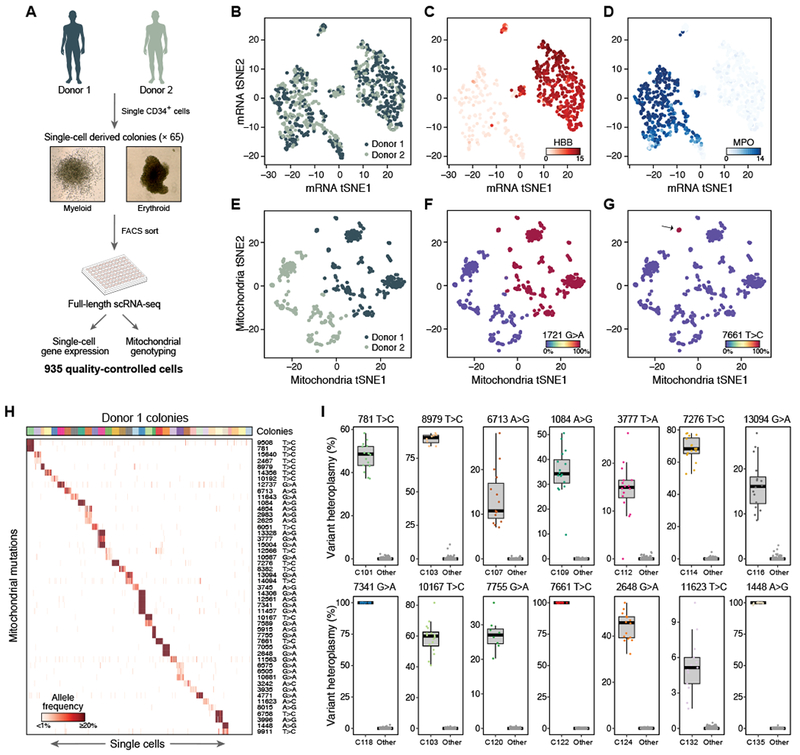
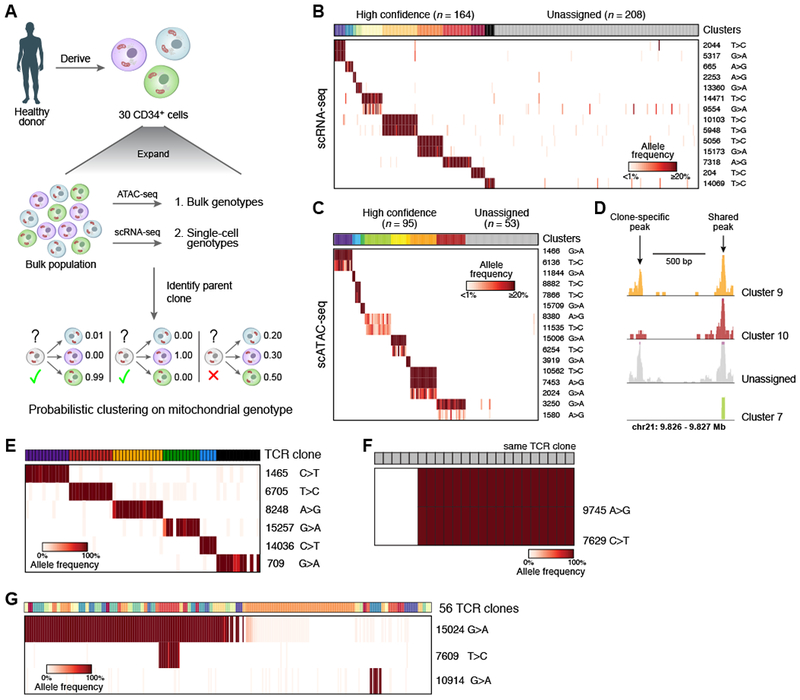
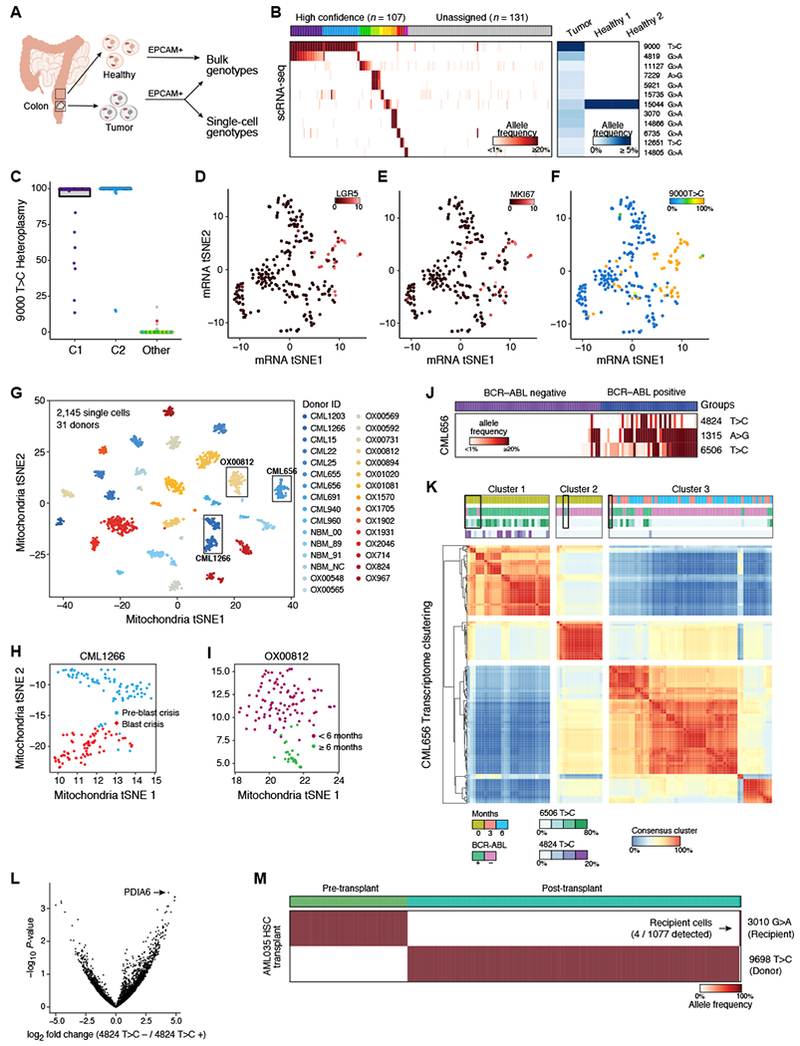
Comment in
-
Mitochondrial barcodes.Nat Methods. 2019 May;16(5):361. doi: 10.1038/s41592-019-0416-9. Nat Methods. 2019. PMID: 31040429 No abstract available.
References
-
- Alemany A, Florescu M, Baron CS, Peterson-Maduro J, and van Oudenaarden A (2018). Whole-organism clone tracing using single-cell sequencing. Nature 556, 108–112. - PubMed
-
- Biasco L, Pellin D, Scala S, Dionisio F, Basso-Ricci L, Leonardelli L, Scaramuzza S, Baricordi C, Ferrua F, Cicalese MP, et al. (2016). In Vivo Tracking of Human Hematopoiesis Reveals Patterns of Clonal Dynamics during Early and Steady-State Reconstitution Phases. Cell Stem Cell 19, 107–119. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
